La beta-talassemia (β-talassemia) o anemia mediterranea, è un gruppo di malattie ereditarie del sangue. Esse sono causate da una ridotta o assente sintesi delle catene beta dell’emoglobina (emoglobinopatia); ciò può comportare risultati variabili, che vanno da una grave anemia a una condizione clinicamente asintomatica.
La talassemia può conferire un grado di protezione contro la malaria, che è, o è stata, diffusa nelle regioni in cui la talassemia è comune, conferendo un vantaggio di sopravvivenza selettiva (noto come vantaggio eterozigote), perpetuando così la mutazione. Per questo motivo è più diffusa nelle aree paludose.
In Italia la maggior incidenza si riscontra in Sardegna.

Le mutazioni che portano ad un deficit di catene β sono varie e sono note diverse delezioni, più o meno estese, del gene, ma le forme più frequenti sono mutazioni puntiformi che interessano la sequenza codificante, la regione del promotore, il codone d’inizio o di stop e le sequenze di splicing. Sono state indentificate circa 200 mutazioni ereditate in modo autosomico recessivo. Il gene per le catene β appartiene ad un cluster genico nella banda 15 del braccio corto del cromosoma 11.
La produzione di catene β può venire a mancare del tutto (β0, come avviene nella mutazione β039, la più diffusa in Italia) oppure può essere ridotta in maniera più o meno marcata (mutazioni di tipo β+, come avviene per alcune varianti che alterano lo splicing o situate nelle regioni 5’ e 3’ del gene).
La produzione di catene β può essere alterata anche in presenza di un gene β intatto, in conseguenza di un’ampia delezione che coinvolge la LCR (Locus control region: Regione di controllo del locus).
Il blocco del gene HBB comporta una diminuzione della sintesi della catena β dell’emoglobina. L’incapacità del corpo di costruire nuove catene porta alla sottoproduzione di HbA (formata da 2 catene alfa e 2 catene beta) in favore della produzione di HbA2, una forma di emoglobina formata da 2 catene alfa e 2 catene delta, invece di beta. La riduzione di HbA totale disponibile a riempire i globuli rossi, a sua volta, porta all’anemia microcitica. L’anemia microcitica sviluppa infine un funzionamento insufficiente dei globuli rossi. L’anemia mediterranea provoca globuli rossi molto piccoli detti microciti che non sono in grado di sintetizzare emoglobina.
I quadri clinici variano in rapporto alla quantità di catene β che l’individuo è in grado di produrre e quindi dalla natura della mutazione. L’omozigote β0/β0 sarà affetto da una forma grave (talassemia maior o anemia di Cooley) che si associa ad anemia microcitica e ipocromica, da diseritropoiesi ed emolisi; mentre gli eterozigoti composti β0/β+ e gli individui β+/β+, mostreranno un quadro clinico più attenuato (talassemia intermedia).
Si parla di talassemia minor o tratto talassemico nel caso dell’eterozigote sano asintomatico oppure oligosintomatico (solo uno degli alleli della β globina porta una mutazione: β+/β; βo/β). È possibile incontrare anche individui doppi eterozigoti, cioè portatori sani sia della forma β che della forma α.

Clinica
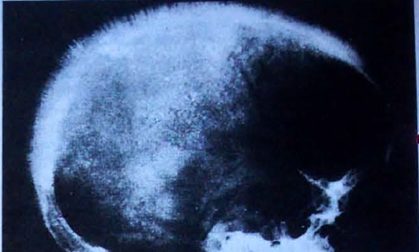
- Il quadro clinico della talassemia maior compare dopo alcuni mesi (6-24 mesi) dalla nascita, quando la progressiva sostituzione dell’emoglobina fetale (α2γ2) con quella adulta (α2β2) rivela il deficit di catene β.
Il segno principale è rappresentato dall’anemia ipocromica microcitica (che determinerà pallore, debolezza e scarsa crescita).
In seguito, i meccanismi di compenso midollare provocano diseritropoiesi con ipertrofia del tessuto emopoietico e deformazioni scheletriche del cranio e delle ossa del volto. La diploe del cranio (tessuto osseo spugnoso ben vascolarizzato, posto tra i tavolati esterno e interno delle ossa piatte del cranio) aumenta di spessore e radiologicamente si evidenzia il cosiddetto “cranio a spazzola”, mentre l’ipertrofia dell’osso malare fa assumere al volto un aspetto “a scoiattolo”.
È presente, inoltre, ipertrofia del fegato e della milza (epatosplenomegalia), che riacquisiscono la funzione di organi emopoietici, ma anche ittero per l’elevata emolisi e litiasi biliare.
Può portare al decesso prima dei 20 anni, di solito per insufficienza cardiaca (siderosi miocardica). - La talassemia intermedia (BTI) raggruppa insieme circa il 10% delle forme omozigoti della malattia e numerose forme eterozigoti composte. L’anemia è variabile, ma è meno grave e viene diagnosticata più tardi. I pazienti affetti da BTI possono necessitare occasionalmente di trasfusioni. Può essere presente ipersplenismo, litiasi biliare, emopoiesi extramidollare, complicazioni trombotiche e progressivo sovraccarico di ferro.
- Le forme eterozigoti (minor o tratto talassemico) di solito presentano alterazioni di laboratorio: pseudopolicitemia ipocromica e microcitica, ma sono asintomatici.

Diagnosi
La diagnosi di β-talassemia si basa sull’analisi dell’Hb con elettroforesi o HPLC (cromatografia liquida ad alta prestazione) e i valori dell’emocromo (MCV < 80 fL, aumento del numero di GR, riduzione di Hb). Nella talassemia maior l’HbA è assente o molto ridotta e l’HbF è predominante.
Gli eterozigoti sono individuati dalle alterazioni all’emocromo (volume globulare medio ridotto e aumento dei GR con Hb normale). L’elettroforesi dell’emoglobina con valori di HbA2 e HbF superiori alla norma fornisce conferma di eterozigosi. Qualora questi parametri non risultino chiaramente significativi, come nel caso di alcune forme eterozigoti, occorre approfondire con indicatori aggiuntivi o con lo studio e sequenziamento del DNA. Questo test è utilizzato per indagare delezioni e le mutazioni nei geni produttori di alfa e beta globina. Studi di famiglia possono essere condotti per valutare lo stato di portatore e il tipo di mutazioni presenti in altri membri della famiglia.
La consulenza genetica è raccomandata per caratterizzare la mutazione, programmare la presa in carico dei bambini affetti e, eventualmente, per la diagnosi prenatale. In caso di diagnosi prenatale di condizioni di omozigosi per l’allele β039 è possibile optare per un’interruzione volontaria della gravidanza.
Terapia e prevenzione

La terapia è trasfusionale per mantenere valori di Hb tra 90-100 g/l, ma l’eccessivo carico di ferro conseguente risulta tossico, in particolare per il cuore (siderosi miocardica), fegato, pancreas e altri tessuti endocrini, con tutta la clinica e sintomatologia conseguente alla disfunzione di tali organi (scompenso cardiaco). Questa complicanza viene contrastata con una terapia ferrochelante (deferoxamina o deferiprone), per infusione o per os e con il controllo del sovraccarico di ferro nei tessuti con la RMN.
L’aspettativa di vita di questi pazienti è comunque ridotta.
In presenza di donatore compatibile può essere preso in considerazione il trapianto di midollo osseo, ma vanno considerati i rischi di questa terapia che possono essere anche mortali. Si utilizzano quindi cellule staminali ematopoietiche provenienti da soggetti istocompatibili o autologhe (dopo correzione del difetto con tecniche d’ingegneria genetica).
Spesso viene rimossa la milza (splenectomia) che risulta ingrandita.
La prevenzione attraverso l’individuazione delle coppie a rischio rappresenta attualmente il modo più efficace di controllo della β-talassemia. Gli eterozigoti sono, di solito, facilmente individuabili dal momento che presentano un volume globulare medio nettamente ridotto rispetto ai valori normali, mentre il numero dei globuli rossi risulta elevato. L’elettroforesi dell’emoglobina con valori di HbA2 e HbF superiori alla norma fornisce conferma di eterozigosi. Qualora questi parametri non risultino chiaramente significativi, occorre approfondire con indicatori aggiuntivi o con lo studio del DNA.
Fonte: Genetica umana e medica.