Per mutazione intendiamo un cambio permanente nella sequenza del gene ereditabile per via germinale.
Le mutazioni sono moltissime e possono interessare uno o più geni.
In base alla porzione di DNA interessata dalla mutazione distinguiamo: mutazioni geniche o puntiformi, che interessano i singoli nucleotidi o pochi nucleotidi; mutazioni cromosomiche, quando interessano parti intere di cromosomi; mutazioni genomiche, quando parliamo di interi cromosomi che vengono perduti o duplicati in maniera non corretta.
Fonte: Le basi patologiche delle malattie.
Le mutazioni geniche si distinguono in:
- Sostituzioni di basi, quando un nucleotide viene sostituito con un altro nucleotide: questo può dar luogo ad un aminoacido diverso;
- Inserzioni, quando in un gene vengono inseriti per sbaglio uno o più nucleotidi; questo, normalmente, provoca uno slittamento della cornice di lettura delle triplette. Quindi, quando questa cornice di lettura viene sfasata per aggiunta di uno o due nucleotidi (o multipli di uno e due), si parla di mutazione frame-shift. Questa, appunto, sposta la cornice di lettura e così la proteina non viene più prodotta in maniera corretta.
- Delezioni, in cui accade la stessa cosa dell’inserzione, la perdita di basi di DNA (uno o due o multipli), sposta la cornice di lettura del DNA. Se abbiamo inserzioni o delezioni di multipli di 3 basi, la cornice di lettura viene invece mantenuta.
Un altro tipo di distinzioni che si può fare è tra mutazioni ereditarie (ereditate in via germinale, che possono portare ad un vantaggio evolutivo; alla morte dell’embrione, soprattutto in caso di aberrazioni cromosomiche o a patologie ereditarie) e mutazioni somatiche che compaiono de novo dopo la nascita (responsabili della comparsa di tumori, della morte cellulare o di patologie congenite).
Per quanto riguarda le sequenze interessate, queste mutazioni possono riguardare una singola sequenza o provocare scambi tra due o più sequenze.

Cause di mutazione
Le cause di mutazioni possono esser spontanee, ovvero legate a quella che è la normale replicazione del DNA, oppure indotte da mutageni.

La causa più comune di mutazione sta nella replicazione del DNA stesso e avviene perciò in maniera spontanea. Considerando che in un uomo adulto c’è un numero elevatissimo di cellule (1014), e che quasi ognuna di queste duplica il proprio DNA, si stima che, nell’intera vita, servono 1025 nuovi nucleotidi, che vengono inseriti per formare nuovo DNA.
L’enzima che replica DNA, la DNA polimerasi, commette circa un errore ogni miliardo di basi incorporate. Quindi, considerando tutto, ogni persona, casualmente, accumula più di mille miliardi di mutazioni nel corso della vita. Questo numero sembra spaventoso, ma in realtà quasi tutto il DNA non è codificante, quindi queste mutazioni avvengono facilmente in regioni in cui non portano a nessun tipo di effetto; allo stesso modo, se la mutazione di un gene che sintetizza per una proteina dell’occhio avviene in una cellula della mano, tale mutazione non darà nessun effetto. Quindi quasi tutte le mutazioni non hanno un effetto a livello fenotipico.
La replicazione cellulare aumenta il rischio di mutazione del DNA perché, replicandolo, il DNA è a rischio di incorporare basi errate. In conformità con ciò, è stato visto che, in malattie genetiche, malattie de novo sono più frequentemente di origine maschile. Infatti, ad esempio, il retinoblastoma, compare de novo in un paziente, all’improvviso in un albero genealogico dove non si era mai presentato, ed è causato nell’88% dei casi dall’ereditarietà paterna. Questo accade perché le cellule uovo subiscono molte meno divisioni degli spermatozoi. La spermatogenesi avviene ogni 16 giorni; quindi, quando uno spermatozoo feconderà una cellula uovo, a seconda dell’età dell’uomo, esso avrà subito un numero più o meno elevato di mutazioni dovute a replicazioni, mentre la cellula uovo matura subisce solo due mitosi subito dopo la maturazione della cellula uovo primaria e non subisce più altre replicazioni di DNA. Questo ci fa capire come l’età del padre sia importante per aumentare il rischio della comparsa di malattie genetiche de novo nei figli.
Oltre alle mutazioni spontanee, vi sono anche tantissimi agenti che aumentano il tasso di mutazioni: agenti chimici, fisici o biologici. Abbiamo:
- agenti esogeni, come ad esempio le radiazioni, che sono capaci di causare danni a livello del DNA provocando rotture a singolo o a doppio filamento; formando dimeri di Timina, che distorcono la struttura primaria del DNA; alchilando alcune basi di DNA, ovvero legando dei gruppi chimici che bloccano la funzione del DNA (tra questi ci sono anche alcuni farmaci antitumorali, che bloccano la crescita delle cellule tumorali).
- agenti endogeni, che causano mutazione nel genoma: eventi che avvengono solitamente nell’individuo. Ad esempio, il differenziamento dei linfociti B e T, si basa su queste mutazioni spontanee, che vengono indotte in maniera tale da ottenere linfociti capaci di riconoscere qualsiasi antigene non-self. Anche i prodotti del metabolismo cellulare generano radicali liberi, che possono andare a cambiare la struttura del DNA, provocando mutazioni. Gli elementi trasponibili (o trasposoni), sono sequenze del DNA che si “traspongono” da una parte all’altra del genoma (non è chiara la loro funzione), e a volte possono causare mutazioni. L’infiammazione è un’altra causa di mutazione. Infine, eventi spontanei che possono avvenire nel DNA, al di là delle mutazioni dovute a replicazione (depurinazioni, deaminazioni).

Mutazioni in DNA codificante
Per quanto riguarda le mutazioni a livello del DNA codificante, quando consistono nella sostituzione di un singolo nucleotide, queste possono essere:
- silenti, quando il nucleotide che viene sostituito dà luogo allo stesso aminoacido (fatto dovuto al codice genetico degenerato);
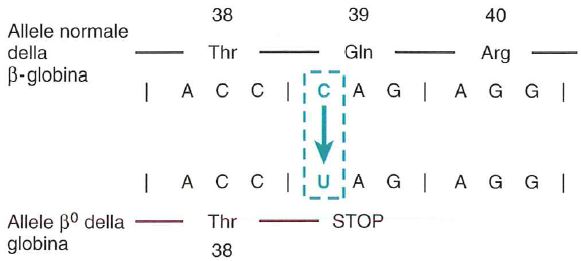
- non-sense, quando viene inserito un codone STOP;
- missenso, quando viene inserito un aminoacido o simile a quello originale (missenso conservativa) o funzionalmente diverso (missenso non conservativa), che quindi darà conseguenze a livello della proteina.
- nei siti di splicing, i siti dove vengono eliminati gli introni: in questo caso si distruggono i segnali di splicing, e così può succedere che un introne venga mantenuto, o che un esone venga rimosso.
Infine, ci sono mutazioni particolari che portano all’amplificazione di triplette nucleotidiche, scoperte più recentemente. Sono malattie ereditarie, ma con ereditarietà particolare: vi sono, in alcune regioni di alcuni geni, alcune triplette che aumentano il proprio numero tra una generazione e l’altra. Il meccanismo non è ben chiaro, ma è probabilmente dovuto a errori di appaiamento del DNA durante la replicazione. Questi nucleotidi, quindi, si “espandono”, fino a portare a delle vere e proprie conseguenze a livello fenotipico. Abbiamo diverse malattie dovute a triplette ripetute, e il numero di ripetizioni può essere molto variabile tra le varie malattie. Generalmente, si parla di pre-mutazione, quando il numero di triplette è al di sotto di una certa soglia; superata quella soglia, si hanno effetti a livello fenotipico. La caratteristica particolare di queste patologie è che la malattia peggiora di generazione in generazione e, andando avanti, compare in età sempre più precoci: il cosiddetto fenomeno dell’anticipo.
La più conosciuta di queste patologie è la sindrome dell’X fragile. In questa patologia la tripletta ripetuta, CGG, è al livello non della sequenza codificante, ma del promotore. Un aumento incontrollato di questa tripletta da una generazione all’altra porta, alla fine, a silenziare il promotore stesso: la proteina, così, non viene più prodotta. In questo caso, sono affetti individui che presentano più di 200 ripetizione della tripletta CGG. Il gene affetto è il gene FMR-1 (familiary mental retardation), perché questa malattia porta a ritardo mentale severo.
Un’altra malattia da espansione di tripletta è la Corea di Huntigton, che riguarda una porzione codificante del genoma: il gene della Huntingtina. La tripletta CAG si espande in maniera incontrollata, sempre da una generazione all’altra, fino a dar luogo a proteine tossiche, che precipitano a livello neuronale, causando, anche in questo caso, deficit a livello neuronale. In questo caso, il numero di triplette di CAG che è un grado di far sì che compaia la malattia è 35: oltre le 35 ripetizioni di questa tripletta compare la malattia.
Nel caso dell’X fragile, la tripletta viene espansa durante l’oogenesi, e quindi è la madre a trasmettere la malattia; nel caso invece della malattia di Huntigton, questa espansione incontrollata di triplette avviene durante la spermatogenesi, quindi è il padre a trasmettere la patologia.

Mutazione in sequenze non codificanti

Per quanto riguarda le mutazioni di sequenze non codificanti, queste possono avvenire a livello degli introni o dei siti di splicing (l’RNA, prima di maturare è formato da esoni ed introni; gli introni vengono rimossi con il meccanismo di splicing, e rimangono solo gli esoni).
Il meccanismo di splicing si basa su due nucleotidi (GU e AG, rispettivamente siti donatori e accettori). Si forma un loop tra G e A (quest’ultima in mezzo all’introne), cade il taglio a livello del sito accettore, e viene rimosso l’introne. Quindi sono molto importanti queste sequenze terminali dell’introne (GU e AG, ma anche l’A mediana). Mutazioni in questi nucleotidi possono portare a splicing anomali.
Se viene mutato un sito a livello di un introne, può succedere che esso venga mantenuto, invece di essere rimosso (ritenzione dell’introne; chiaramente in questo modo la sequenza di mRNA cambia completamente, perché verranno inseriti aminoacidi che non c’entrano niente con la sequenza originale);
ci può essere anche la perdita di un esone (vengono rimossi due introni che fiancheggiano un esone, ma anche l’esone centrale; così viene deleta un’intera porzione di mRNA); oppure ci sono alcune mutazioni puntiformi che possono formare i cosiddetti siti criptici di splicing: se una mutazione fa in modo che si venga a creare un sito donatore GU o un sito accettore AG, ci può essere un nuovo splicing non previsto precedentemente, e quindi abbiamo una situazione intermedia, in cui vengono rimosse una porzione di introne e una porzione di esone. Anche in questo caso, comunque, cambia la sequenza finale di mRNA.
Fonte: Genetica umana e medica.