L’ipercolesterolemia familiare è una malattia genetica con ereditarietà autosomica dominante, cioè basta una copia del gene mutato perché la patologia si manifesti.
In eterozigosi la patologia è molto diffusa, ha una prevalenza (incidenza) di 1 su 500. È responsabile del 5% degli infarti prima dei 60 anni ed è caratterizzata da un livello di colesterolo 2 volte il livello normale e cioè 400 mg.
Il difetto riguarda il gene per il recettore delle LDL (Low Density Lipoprotein). I pazienti eterozigoti presentano xantomi che sono depositi giallastri di colesterolo presenti nei tendini e cute, l’arcus corneale in cui ci sono depositi giallastri di colesterolo alla periferia della cornea ed aterosclerosi con riduzione del diametro dei vasi responsabile di coronaropatia, di infarto miocardico e di ictus cerebrale.

In omozigosi la patologia è molto rara, l’incidenza è di 1 su 1.000.000 e la quantità di colesterolo è incompatibile con la vita ed è >600 mg fino a 1.000 e arriva a 6 volte il livello normale di colesterolo; gli omozigoti infatti hanno infarto miocardico prima dei 20 anni. Vi sono modificatori genetici come il sesso maschile che è più a rischio rispetto a quello femminile, particolari gruppi etnici che sono più a rischio e modificatori ambientali quali la dieta, l’attività fisica e farmaci ipocolesterolemizzanti.
Lo xantoma è una degenerazione della pelle di color giallastro dovuto ad accumulo di lipidi. Si riscontra particolarmente in caso di livelli elevati di colesterolo e di trigliceridi. Gli xantomi possono essere localizzati anche a livello dei tendini. Il termine deriva dal greco xanthos che significa giallo.
Con l’aterosclerosi si ha restringimento del lume dei vasi perché le LDL sono catturate dai macrofagi che hanno i recettori per le LDL, detti recettori scavenger. Questi recettori si infarciscono di lipoproteine e diventano cellule schiumose dando origine al processo infiammatorio. I macrofagi attaccando le LDL innescano un processo infiammatorio e stimolano le cellule muscolari lisce della tonaca media a proliferare. Le cellule lisce stimolate producono una matrice extracellulare che restringe il lume del vaso e in questo modo si forma il cappuccio fibroso che riduce il lume del vaso (ateroma).

Fonte: Le basi patologiche delle malattie.
Turnover delle lipoproteine
Il 7% del colesterolo circola nel plasma sotto forma di LDL. Quando le LDL sono escrete dal fegato si chiamano VLDL (Very Low Density Lipoprotein) lipoproteine a bassissima densità e presentano oltre a esteri del colesterolo e trigliceridi 3 apoproteine: ApoC, B-100 e ApoE. Quando queste particelle penetrano nei vasi sanguigni e arrivano alle cellule muscolari e alle cellule adipose, dove i trigliceridi vengono lasciati sotto forma di deposito e l’apoC viene rimossa, le particelle sono chiamate IDL (lipoproteine a densità intermedia). Poi continuano a perdere trigliceridi e apoproteine e acquistano ancora colesterolo, resta solo la B-100 e diventano LDL.
Il recettore per le LDL del fegato è in grado di captare sia LDL che IDL. È proprio una mutazione di questo recettore a livello epatico ad essere responsabile dell’ipercolesterolemia familiare.
Dopo che si legano al recettore per LDL si formano le fossette rivestite che sono invaginazioni della membrana, poi le vescicole rivestite che si fondono coi lisosomi, che sono ricchi di enzimi che scindono il colesterolo dalle porzioni proteiche (B-100), che servono a sintetizzare amminoacidi. Il colesterolo rimanente nella cellula serve per la fluidità della membrana cellulare, come base per gli ormoni steroidei e per gli acidi biliari.

Fonte: Le basi patologiche delle malattie.
Se è prodotto in eccesso, vi è un meccanismo a feed-back negativo per cui il colesterolo inibisce l’idrossimetil glutaril-CoA reduttasi che blocca la sintesi di colesterolo, inoltre l’eccesso di colesterolo stimola l’acetil-CoA che è importante per l’immagazzinamento del colesterolo; inoltre il colesterolo inibisce nel DNA, a livello di alcuni fattori di trascrizione, la sintesi dei recettori per LDL, cioè se vi è già colesterolo dentro la cellula è inutile portarne dentro altro.

Classi di mutazione nel gene del recettore delle LDL
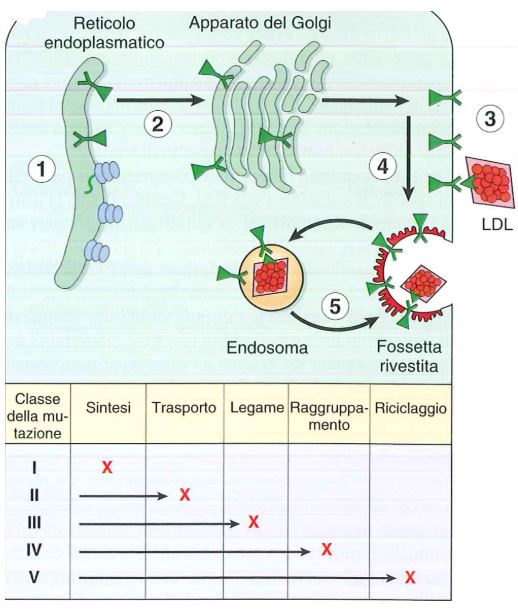
Fonte: Le basi patologiche delle malattie.
Vi sono tante mutazioni che colpiscono il gene del recettore LDL, classificate in 5 classi:
- Alla classe 1 appartengono le mutazioni che impediscono la sintesi del recettore (sono mutazioni che inseriscono i codoni di stop e mutazioni frame shift);
- Le mutazioni di classe 2 sono mutazioni che sostituiscono amminoacidi, sono mutazioni missenso, che rendono le proteine anomale per cui il recettore non arriva in membrana in quanto viene degradato prima a livello del Golgi;
- Le mutazioni di classe 3 riguardano i domini di legami del recettore LDL, cioè il recettore riesce ad arrivare in membrana ma non riesce a legare le LDL;
- Classe 4: il recettore è capace di legare le LDL ma è impedita l’invaginazione del recettore per cui rimane in superficie ma non riesce a internalizzare le LDL che restano all’esterno;
- Classe 5: sono le meno gravi, non c’è regolazione del riciclo del recettore, questo è invaginato però vi sono errori che impediscono al recettore di tornare in membrana.
[text-blocks id=”14550″ slug=”banner-adsense”]