Il metodo del sequenziamento di proteine fu messo a punto da parte dello scienziato F. Sanger.
La determinazione della sequenza primaria della catena polipeptidica attualmente viene fatta in maniera automatica in modo molto veloce da strumenti abbastanza complessi chiamati sequenziatori (automatici) che sono presenti disponibili in vari laboratori, mentre nel 1958, quando è stato messo a punto questo metodo da Sanger, ci volevano parecchi anni per sequenziare una proteina.
È possibile conoscere la composizione amminoacidica di una proteina, ossia il tipo e il numero di amminoacidi che costituiscono la proteina, andando a denaturare completamente la proteina. Le condizioni altamente denaturanti sono quelle che si ottengono in ambiente fortemente basico o fortemente acido facendo bollire la proteina in una soluzione 6 M di HCl, che è una concentrazione altissima di acido, in questo modo i singoli legami peptidici che tengono insieme l’amminoacido vengono rotti, queste sono condizioni estreme ed è chiaro che la proteina non solo è denaturata, ossia perde la sua conformazione, ma si ha l’idrolisi di tutti i legami peptidici e la proteina viene rotta nelle sue parti costitutive. In questo modo otteniamo una miscela di amminoacidi che costituisce la nostra proteina di interesse e viene analizzata con una tecnica chiamata HPLC (high performance liquid cromatografy) oppure con una cromatografia a scambio ionico.

Queste sono delle tecniche cromatografiche che permettono di determinare la tipologia degli amminoacidi e quindi di risalire al numero ma soprattutto al tipo di amminoacidi che costituiscono la nostra proteina. Tuttavia in questo modo, avendo frammentato la proteina, non potremmo mai conoscere l’ordine con cui sono legati e quindi ricostruire la nostra proteina.
Un altro tipo di metodo permette di marcare l’amminoacido N-terminale facendolo reagire con un marcativo che si chiama 1 fluoro 2,4 dinitrobenzene (FDNB) anche noto come reattivo di Edman. Infatti il metodo di sequenziamento delle proteine che va sotto il nome di Sanger può essere chiamato anche degradazione di Edman, scienziato che ha scoperto e messo in evidenza la capacità di far reagire FDNB con il gruppo amminico dell’amminoacido N-terminale che è l’unico a presentare il gruppo amminico libero. Successivamente in ambiente acido (6 M HCl) si ha la rottura dei legami peptidici, compreso quello dell’amminoacido legato al FDNB permettendo di evidenziarlo.
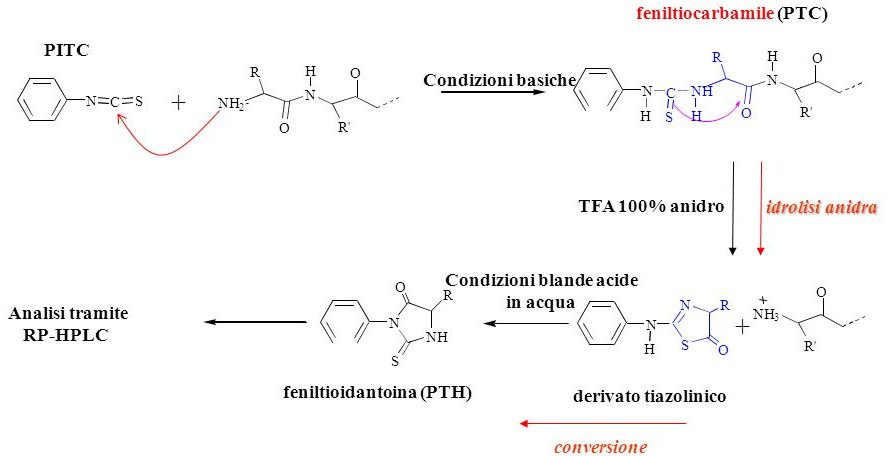
Il passo successivo è stato quello di utilizzare il PTC (fenilisotiocianato), un altro reattivo che in ambiente basico crea un addotto legandosi con la catena polipeptidica. Poi si tratta il mix di reazione variando il pH da basico a acido si ottiene la rottura del legame peptidico con la formazione dell’addotto più stabile con il PTC formando il PTH (feniltioidantoina). Quindi a pH basico abbiamo il legame tra il gruppo amminico sul carbonio del PTC, si formano questi due metaboliti intermedi e cambiando il pH da basico a acido otteniamo la rottura dell’amminoacido N-terminale con la formazione di PTH.
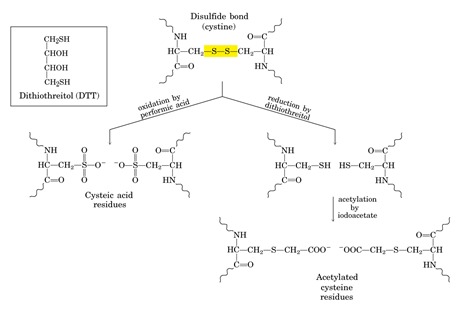
A questo dobbiamo aggiungere delle operazioni che precedono il sequenziamento di una proteina, perché quando andiamo a sequenziare una proteina, dobbiamo tener conto anche della sua struttura terziaria e quaternaria se presente, quindi dobbiamo ottenere le singole catene polipeptidiche andando a sequenziare una per volta. Si procede allora con la rottura del ponte disolfuro in ambiente acido ad esempio con l’acido performico oppure trattando la proteina con un agente molto basico che è il DTT (ditiotreitolo) oppure il β-SH (β-mercaptoetanolo). Questi ultimi due sono caratterizzati dalla presenza di gruppi tiolici che sono in grado di ridurre il ponte disolfuro ossia riportarlo allo stato ridotto, di gruppi tiolici singoli.
Quando si utilizza l’acido performico, il ponte di solfuro viene rotto formando residui di acido cisteico che impediscono ai gruppi solforati di ricongiungersi a formare il ponte disofuro.
Caricamento….

La reazione con il PTC può essere ripetuta fino a 60 volte e permette di determinare la sequenza primaria di frammenti di una proteina non molto lunghi fino a 40-60 residui amminoacidici, perché a un certo punto la reazione non può andare avanti, si accumulano i vari metaboliti che vengono prodotti e la reazione viene stoppata. Questo vuol dire che dato che ci sono moltissime proteine che sono formate da più di 60 frammenti allora la proteina prima di essere sottoposta a sequenziamento viene frammentata in vari segmenti di grandezza al di sotto di 40-60 monomeri e questo si può fare con gli enzimi digestivi come la tripsina che rompe i legami peptidici in corrispondenza di arginina (R) e della lisina (K) e poi questi brevi segmenti vengono sottoposti a reazione con PTC, rilevamento del segnale del singolo amminoacido con le tecniche cromatografiche e così si ottiene la sequenza di questi frammenti. Ora il problema è metterli in ordine.
Tramite trattamento con FDNB possiamo identificare in maniera univoca l’amminoacido N-terminale. Per poter mettere in ordine i vari frammenti che noi abbiamo ottenuto con il taglio proteolitico della tripsina utilizziamo un altro agente di taglio, ossia facciamo la stessa operazione di taglio della stessa proteina, però con un altro agente che può essere un altro enzima proteolitico. Il trucco è quello di tagliare la stessa proteina in punti diversi e sequenziare questi frammenti ottenuti da questa seconda reazione in maniera tale che i frammenti che noi otteniamo con questo secondo reagente siano sovrapponibili rispetto agli altri, cioè quando viene sequenziato troveremo dei tratti che sono in comune, un overlapping della sequenza e in questo modo in maniera univoca possiamo andare a metterli in ordine e ottenere la sequenza completa e precisa di questa proteina, dal N-terminale che noi abbiamo identificato con FDNB fino a C-terminale che sarà l’amminoacido finale della nostra sequenza polipeptidica.
Le sostanze chimiche che possono essere utilizzate per il sequenziamento sono: la tripsina che taglia in corrispondenza di K (lisina) ed R (arginina), la proteasi sottomascellare, un enzima non umano, che rompe in corrispondenza di R, oppure la chimotripsina o la proteasi V8 dello stafilococco aureo o la proteasi N-aspartato, la pepsina, l’endoproteinasi lisina C, oppure il bromuro di cianogeno che è l’unico che non è un enzima, cioè una proteina.
Now loading…