La leucemia linfatica cronica (LLC) è una neoplasia caratterizzata dalla proliferazione e dall’accumulo di linfociti B non immunocompetenti. Si manifesta con l’invasione dei linfociti nel sangue periferico e midollare, ma ha una bassa aggressività. È la forma più comune di leucemia nei Paesi occidentali e colpisce principalmente gli adulti (media di 65 anni), con una leggera prevalenza negli uomini. La causa esatta è sconosciuta.
Segni clinici
La LLC può essere asintomatica nel 70% dei casi, rappresentando una linfocitosi casuale. Le manifestazioni cliniche della malattia sono causate dall’infiltrazione progressiva del midollo osseo, dei linfonodi e di altri tessuti da parte dei linfociti, oltre a alterazioni immunologiche. I sintomi comuni includono:
- Sintomi B come febbre, calo ponderale e sudorazione notturna.
- Sindrome anemica che può essere causata da infiltrazione, anemia emolitica autoimmune (Coombs +) o ipersplenismo.
- Infezioni, principalmente batteriche ma anche da herpes virus e germi opportunistici, principale causa di morte, dovute ad alterazioni dell’immunità (ipogammaglobulinemia).
- Trombocitopenia causata da infiltrazione o autoimmune.
- Adenopatie bilaterali e simmetriche.
- Splenomegalia ed epatomegalia.
- Infiltrazione di altri tessuti come la cute, il rene e i polmoni (raro).
- Maggiore rischio di sviluppare seconde neoplasie come il carcinoma della pelle, dell’intestino e dei polmoni, oltre a fenomeni autoimmuni.
Now loading…
Dati di laboratorio e diagnosi
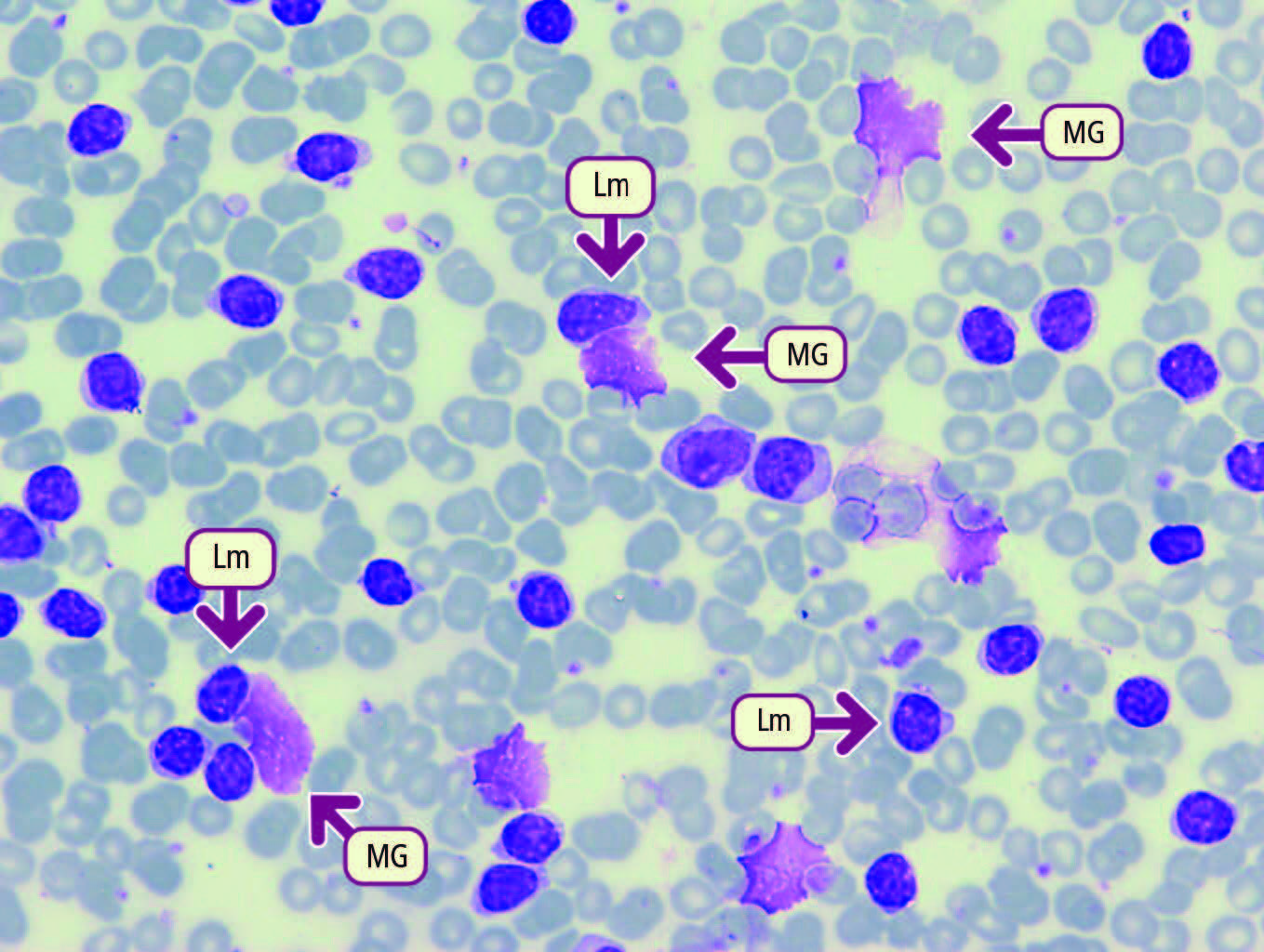
Fonte: Manuale di ematologia SSM.
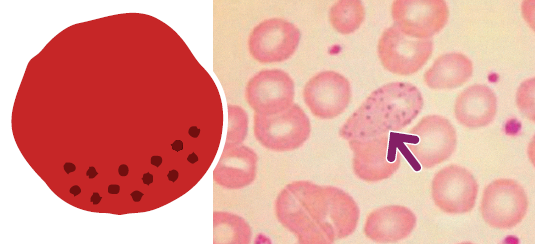
- Emocromo e striscio di sangue periferico mostrano leucocitosi, linfocitosi (linfociti di piccole dimensioni e aspetto maturo con ombre di Gümprecht), anemia e trombocitopenia.
- L’esame del midollo osseo non è richiesto per la diagnosi, ma può mostrare infiltrazione di diverso grado da parte di piccoli linfociti, con quattro possibili pattern (nodulare, diffuso, interstiziale e misto nodulo-interstiziale).
- La citogenetica rivela alterazioni nell’80% dei casi, come del (13q), trisomia 12 o, meno frequentemente, del (17p) che conferisce resistenza alla chemioterapia.
- L’immunofenotipo evidenzia linfociti B (CD19+, CD22+) con coespressione di CD5, CD23 e CD20 (debole).
Altri marcatori includono ipogammaglobulinemia, LDH elevata, test di Coombs diretto positivo e livelli aumentati di β2-microglobulina. Di solito, non è presente una banda monoclonale nel sangue poiché le cellule della LLC non secernono immunoglobuline.
Si riscontra solitamente una conta di linfociti ≥5000/μL con immunofenotipo: CD19+, CD20+ (debole), CD5+ e CD23+.
N.B.: la LLC non rientra nella diagnosi differenziale delle pancitopenie.
Stadiazione
| BINET A | ≤ 2 aree linfonodali* interessate |
|---|---|
| BINET B | ≥ 3 aree linfonodali* interessate |
| BINET C |
| RAI 0 | Linfocitosi nel sangue periferico e nel midollo osseo | Basso rischio |
|---|---|---|
| RAI 1 | Linfocitosi + adenopatie | Rischio intermedio |
| RAI 2 | Linfocitosi + spleno- e/o epatomegalia | |
| RAI 3 | Linfocitosi + Hb <11 g/dL | Alto rischio |
| RAI 4 | Linfocitosi + piastrine <100 × 109/L |

Trattamento
La maggior parte dei pazienti asintomatici non viene trattata e viene sottoposta a monitoraggio periodico.
Il trattamento è giustificato solo se sono presenti segni o sintomi correlati alla malattia, come sintomi B, adenopatie progressive o classificazione di Binet B o C.
Le opzioni di trattamento includono:

- Chemioterapia in combinazione con rituximab o obinutuzumab (anti-CD20) per pazienti giovani senza mutazioni p53 o del (17p).
- Ibrutinib (inibitore della tirosin chinasi di Bruton) per pazienti di tutte le età con mutazioni p53 o del (17p). Nei pazienti giovani, è consigliabile effettuare uno screening per un potenziale donatore allogenico sin dalla diagnosi.
- Clorambucil (alchilante) con o senza rituximab o obinutuzumab per pazienti non idonei con mutazioni p53 o del (17p).
- Seconda linea di trattamento con venetoclax (anti-bcl2) e rituximab, idelalisib (anti-PI3K-delta) o ibrutinib (se non usato precedentemente).
- Ulteriori linee di trattamento possono includere l’uso di CAR-T anti-CD19.
Evoluzione
La LLC può evolvere in una leucemia prolinfocitica con più del 55% di prolinfociti nel sangue periferico, che ha una prognosi infausta, o nella sindrome di Richter con trasformazione in un linfoma a grandi cellule ad alto grado. La maggior parte dei pazienti che sviluppano la malattia in giovane età muore a causa della neoplasia stessa e dell’immunodeficienza umorale. Al contrario, i pazienti che sviluppano la malattia in età avanzata con un decorso indolente tendono a mantenere la loro aspettativa di vita non ridotta.
Now loading…