Il feocromocitoma e il paraganglioma sono malattie rare, con un’incidenza nella popolazione generale di 2-8 casi per milione di abitanti all’anno. La prevalenza nei pazienti ipertesi è dello 0,2-0,4%. Queste patologie sono più frequenti nella quarta e quinta decade di vita, senza differenze significative tra i sessi.
Si tratta di tumori neuroendocrini (NET) derivati dalla cresta neurale, capaci di produrre catecolamine. Sono benigni nell’85-90% dei casi.
In base all’origine, si distinguono in:
- feocromocitoma surrenalico (80-85% dei casi), originato dalla midollare del surrene;
- feocromocitoma extrasurrenalico o paraganglioma funzionante (15-20% dei casi), originato dai gangli associati al sistema nervoso simpatico, localizzati in sede para-aortica/pericavale a livello addominale o mediastinico, o in corrispondenza della biforcazione dell’iliaca (organo dello Zuckerkandl). Più raramente, si trova nel tessuto perineale, nella vescica o nell’uretere;
- paragangliomi non funzionanti, originati dai gangli parasimpatici, generalmente localizzati a livello del collo (glomo carotideo e gangli vagali) e/o della testa (gangli del bulbo giugulare o giugulo-timpanici).
In circa il 20% dei casi, il feocromocitoma è presente nel contesto di sindromi ereditarie come MEN 2, Von Hippel Lindau (VHL), neurofibromatosi tipo 1 (NF1) e sindrome del paraganglioma/feocromocitoma (SPG 1, SPG 4).

Manifestazioni cliniche

Il feocromocitoma è noto come “il grande mimo” per la varietà di segni e sintomi con cui si presenta, spesso aspecifici e riscontrabili in numerose altre condizioni cliniche. L’ipertensione arteriosa è il sintomo più caratteristico, presente nel 90% dei casi, e può manifestarsi sia in forma parossistica (30% dei casi) sia in forma stabile (60% dei casi). Nel 10-50% dei casi si osserva ipotensione ortostatica.
La triade sintomatologica classica comprende cefalea (60-90% dei casi), cardiopalmo (50-70% dei casi) e sudorazione (55-75%). Questi sintomi, associati a crisi ipertensiva e pallore cutaneo, sono dovuti alla brusca liberazione delle catecolamine da parte del tumore e dovrebbero immediatamente indurre al sospetto di feocromocitoma, anche se spesso molti pazienti con “crisi” non hanno tale patologia. La frequenza e la durata delle crisi sono variabili (da pochi secondi ad alcune ore).
Altri segni clinici tipici includono ansietà, flushing, nausea, vomito, astenia, calo ponderale, iperglicemia. Manifestazioni meno frequenti ma di interesse internistico sono dolore toracico o addominale, stipsi (megacolon), ematuria (in caso di localizzazione vescicale), febbre, stato flogistico.
Può presentarsi anche con segni/sintomi cardiaci: angina, infarto (tipicamente a coronarie indenni), aritmie ipercinetiche, cardiopatia ipertrofica o congestizia, edema polmonare acuto, shock. In caso di voluminosi paragangliomi non secernenti, la prima manifestazione clinica può essere rappresentata da sintomi neurologici da compressione.
Il feocromocitoma può essere asintomatico, specie nel contesto di forme familiari (MEN 2, VHL, NF1, SPG) e in caso di incidentaloma surrenalico.
Crisi parossistiche (con bruschi rialzi pressori e aritmie talvolta fatali) possono essere scatenate da manovre endoscopiche, cateterismi e procedure angiografiche con mdc, anestesia, farmaci (beta-bloccanti, metoclopramide, antidepressivi triciclici, metil-DOPA), stimolanti (caffeina, nicotina, teofillina), alcol e alimenti contenenti tiramina (ad esempio cioccolato).
Caricamento…
Aspetti biochimici
Il feocromocitoma è caratterizzato da un’aumentata secrezione di catecolamine [adrenalina (A) e/o noradrenalina (NA), raramente dopamina (DA)] e dei loro metaboliti (metanefrine e acido vanilmandelico, VMA). Generalmente, il feocromocitoma produce sia A sia NA, con prevalenza della seconda. La secrezione prevalente o isolata di A è rara ed è tipica di forme surrenaliche, specie dei casi associati a MEN 2. Il feocromocitoma associato a sindrome VHL può produrre esclusivamente NA. Estremamente rari sono i casi secernenti DA, secrezione più frequentemente associata a un fenotipo maligno.
Valori aumentati di catecolamine si riscontrano frequentemente in condizioni fisiopatologiche di iperattività del sistema simpatico (condizioni di stress, ipoglicemia, freddo, IMA, ictus, interventi chirurgici, broncopneumopatie croniche ostruttive, insufficienza renale cronica) e per cause iatrogene (oltre a quelle già citate prima, clozapina, calcio-antagonisti – somministrazione acuta – vasodilatatori, nitroglicerina, inibitori MAO, cocaina, furosemide, aspirina, eritromicina).
Il feocromocitoma può essere maligno in una percentuale superiore al 10% dei casi. Fattori predittivi di malignità sono le dimensioni (>5 cm), la localizzazione extrasurrenalica, il marcato incremento dei livelli di cromogranina A e di VMA o di DA, la familiarità per neoplasie maligne extrasurrenaliche. Spesso non è possibile predire l’evoluzione maligna sulla base dell’esame istologico: gli unici criteri certi di malignità sono la presenza di metastasi a distanza o l’invasione locale, mentre caratteristiche suggestive sono la nodularità “grossolana”, la necrosi confluente con assenza di corpi ialini, le atipie cellulari con alto indice mitotico (>10 mitosi/mm²) e un elevato indice proliferativo (Ki-67 >10%). Un’elevata probabilità di malignità è descritta nei feocromocitomi/paragangliomi con mutazioni di SDH-B.
Diagnosi
La diagnosi del feocromocitoma è principalmente biochimica, mentre gli esami strumentali sono utilizzati per localizzare il tumore una volta confermata la diagnosi. Prima di procedere ai test diagnostici, è fondamentale sospendere eventuali farmaci interferenti.
Il dosaggio delle metanefrine, in particolare quelle plasmatiche, è più sensibile rispetto a quello delle catecolamine per la diagnosi del feocromocitoma. Le metanefrine sono prodotte costantemente dal tumore e rilasciate nel sangue indipendentemente dalle catecolamine, che invece vengono rilasciate in modo intermittente e in quantità limitate. Pertanto, il test iniziale dovrebbe includere il dosaggio delle metanefrine plasmatiche libere o urinarie, o entrambe se possibile. Non c’è ancora consenso su quale sia il test migliore nella fase di screening, se il dosaggio plasmatico o quello urinario.
Le metanefrine plasmatiche libere frazionate (metanefrina, normetanefrina) sono il test più sensibile, ma i falsi positivi non sono infrequenti e attualmente il loro dosaggio è ancora poco disponibile. Il dosaggio delle metanefrine urinarie è meno sensibile, ma ha un’elevata specificità (99,7%). Questo test dovrebbe essere utilizzato nello screening dei pazienti a basso rischio (sintomatici, con flushing, ipertensione non controllata, incidentaloma surrenalico). È importante considerare la possibilità di falsi negativi (ad esempio riduzione della clearance renale, raccolta urinaria incompleta) e di falsi positivi (ad esempio condizioni di stress e interferenza da parte di farmaci, in particolare β-bloccanti).

Nella pratica clinica sono ampiamente utilizzate anche le catecolamine plasmatiche e urinarie. Per aumentare la specificità dello screening, si può associare alle metanefrine frazionate plasmatiche il dosaggio della cromogranina A (immagazzinata e cosecreta con le catecolamine).
Il dosaggio di acido vanilmandelico urinario (VMA) non dovrebbe essere utilizzato come test iniziale perché poco sensibile (falsi negativi nel 40% dei casi); ha una buona specificità (99% nelle forme familiari), ma il suo impiego è diminuito negli ultimi anni, dopo l’introduzione nella pratica clinica delle metanefrine urinarie.
I test di stimolo sono sconsigliati per il rischio di indurre crisi ipertensive, mentre nei casi dubbi può essere indicata l’esecuzione del test alla clonidina con valutazione della soppressione di catecolamine/metanefrine plasmatiche. Valori francamente elevati di catecolamine/metanefrine (>4 volte il limite di normalità) sono diagnostici per feocromocitoma, ma spesso è indicato un adeguato approfondimento diagnostico in ambito specialistico.
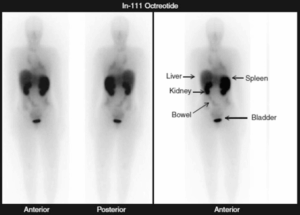
Per localizzare il tumore, si eseguono TC o RMN dell’addome; in caso di negatività, è utile estendere l’indagine a collo, torace e pelvi. La RMN è superiore alla TC nell’individuare le forme extrasurrenaliche e le forme surrenaliche nel contesto di sindromi familiari; inoltre, può essere utilizzata come esame iniziale in età pediatrica, in caso di gravidanza o allergia a m.d.c. La scintigrafia con 123I-MIBG è un esame funzionale dotato di elevata specificità (quasi il 100%) in grado di individuare localizzazioni multiple o metastatiche, ma non c’è consenso su quando debba essere utilizzata. Potrebbe non essere necessaria in caso di tumore surrenalico <5 cm con significativo incremento delle catecolamine.
In casi selezionati, nella diagnosi di sede possono essere utilizzati: Octreoscan (positivo in meno del 50% dei casi), 18F-DOPA PET, FDG-PET e cateterismo venoso.
Genetica
Le principali forme ereditarie di feocromocitoma sono rappresentate dalla sindrome di von Hippel-Lindau e dalla MEN tipo 2, trasmesse con carattere autosomico dominante. Il feocromocitoma può essere presente, anche se raro, nella neurofibromatosi tipo 1, mentre è aneddotico nella MEN tipo 1 e nella sindrome di Carney. Recentemente sono state descritte forme familiari di paraganglioma associato al feocromocitoma (sindromi PGL-1 e PGL-4), anch’esse a trasmissione autosomica dominante, caratterizzate da mutazioni nei geni che codificano per le subunità dell’enzima succinato-deidrogenasi D e B (SDHD e SDHB). Mutazioni nel gene che codifica per la subunità C della SDH (SDHC) sono state sinora riportate solo in caso di paragangliomi associati al sistema parasimpatico.
Sono attualmente disponibili test genetici per la ricerca di mutazioni nei geni RET (correlato alla sindrome MEN II A e MEN II B), VHL (correlato alla sindrome di von Hippel-Lindau), SDHB e SDHD (correlati alla sindrome del paraganglioma/feocromocitoma). A seconda del diverso contesto sindromico, il feocromocitoma può assumere connotazioni cliniche diverse, che implicano modalità differenziate di screening e follow-up. Rispetto a quello sporadico, il feocromocitoma familiare insorge in età più precoce, può essere bilaterale o multifocale, tende a recidivare con maggior frequenza e può avere fenotipo biochimico caratteristico (ad esempio il feocromocitoma associato alla sindrome VHL produce quasi esclusivamente NA, per ridotta espressione dell’enzima fentolamina N-metiltransferasi, che converte la NA in A).
I test genetici sono consigliati nei pazienti con familiarità per feocromocitoma, ma devono essere presi in considerazione anche nei soggetti con forma sporadica, specie se di età inferiore ai 50 anni, se con tumore multifocale, maligno o bilaterale.
Caricamento….
Terapia
Il trattamento di scelta per il feocromocitoma è l’asportazione chirurgica del tumore. Tuttavia, per ridurre la mortalità, è indispensabile un team esperto di chirurghi e anestesisti, oltre a una gestione pre, intra e post-operatoria adeguata da parte degli specialisti. Il tipo di intervento deve considerare il tipo di tumore, la sede, le dimensioni e il background familiare. L’approccio laparoscopico è di prima scelta per il feocromocitoma o il paraganglioma addominale. Nei casi di feocromocitoma ereditario, spesso bilaterale, può essere presa in considerazione una procedura chirurgica con risparmio della corteccia surrenalica per prevenire un ipocorticosurrenalismo permanente, ma va tenuto in considerazione l’aumentato rischio di recidive connesso a questo trattamento.
Le complicazioni intra-operatorie includono crisi ipertensive, aritmie cardiache, edema polmonare e ischemia cardiaca, dovute al brusco rilascio di catecolamine durante l’intervento, che possono essere fatali. Nel post-operatorio, si possono verificare ipoglicemia, grave ipotensione e shock per la brusca caduta dei livelli di catecolamine circolanti in presenza di farmaci α- e β-bloccanti.
Preparazione Farmacologica

La preparazione farmacologica all’intervento deve essere iniziata almeno 10-14 giorni prima e prevede l’utilizzo di un α-bloccante (generalmente doxazosina) e di un calcio-antagonista in caso di ipertensione non controllata. Il β-bloccante (propranololo o atenololo) può essere introdotto solo alcuni giorni dopo il blocco degli α-recettori ed è particolarmente utile nei pazienti con tachiaritmia. È importante ricordare che l’uso del β-bloccante non deve mai essere iniziato prima dei farmaci α-bloccanti, poiché la marcata vasodilatazione mediata dai recettori β-adrenergici porta a un’eccessiva ed incontrastata stimolazione dei recettori α, con conseguenti crisi ipertensive. Inoltre, per ridurre il rischio di ipotensione ortostatica, è utile una dieta con supplementazione di sodio e la somministrazione di liquidi.
In corso di crisi ipertensiva possono essere utilizzati urapidil e nitroprussiato sodico; in caso di aritmia, lidocaina ed esmololo (β-bloccante a breve emivita per aritmie sopraventricolari). In alcuni casi con ipertensione e aritmie non controllate da α- e β-bloccanti, è stata utilizzata anche l’alfa-metil-para-tirosina, un inibitore della sintesi delle catecolamine, ma al momento non vi sono studi clinici prospettici randomizzati che stabiliscano quale sia il trattamento pre-operatorio più efficace.
Follow-up
Il follow-up deve essere protratto per molti anni a causa della possibilità di recidive in altre sedi (descritte in oltre il 10% dei casi, anche dopo 10 anni dall’intervento chirurgico) o di metastasi in caso di malignità. Quando viene confermata la malignità, il decorso clinico è molto variabile, con una sopravvivenza media a 5 anni del 50%.