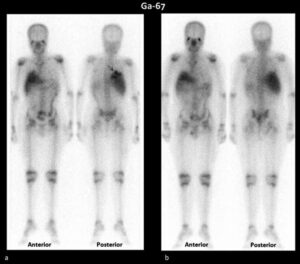
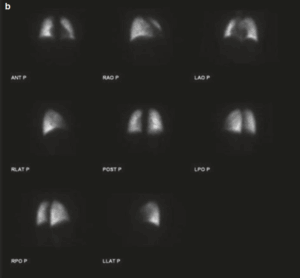
La sarcoidosi si definisce come una patologia infiammatoria multisistemica di natura cronica, la cui manifestazione patognomonica risiede nella formazione di granulomi epitelioidi non caseosi in molteplici organi e tessuti.1 2
Sebbene la presentazione clinica mostri una straordinaria eterogeneità, il coinvolgimento dell’apparato respiratorio, in particolare dei polmoni e dei linfonodi intratoracici, si verifica in oltre il 90% dei pazienti adulti, rendendo la gestione pneumologica il cardine del percorso assistenziale.
La natura enigmatica della malattia risiede in un’eziologia ancora non del tutto chiarita, che sembra scaturire da una risposta immunitaria aberrante e persistente innescata da trigger ambientali o infettivi in soggetti caratterizzati da una specifica vulnerabilità genetica.3
Indice
Caricamento…
- Caratterizzazione istopatologica
- Architettura epidemiologica e varianti demografiche
- Eziopatogenesi
- Suscettibilità genetica
- Trigger ambientali e infettivi
- Meccanismi immunologici della granulomatosi
- Reazioni sarcoidee indotte da farmaci e biotecnologie (DISR)
- Inibitori dei Checkpoint Immunitari (ICI)
- Antagonisti del TNF-alfa
- Clinica
- Apparato respiratorio (>90%)
- Manifestazioni cutanee (25%)
- Coinvolgimento oculare (12-25%)
- Sarcoidosi cardiaca e neurologica
- Percorso diagnostico e stratificazione del rischio
- Stadiazione radiografica di Scadding
- PET-TC con FDG
- Biopsia e tecniche endoscopiche
- Laboratorio e altre indagini
- Diagnosi differenziale e identificazione dei “Mimics”
- Paradigmi terapeutici e Linee Guida Internazionali
- Terapia di prima linea: glucocorticoidi
- Terapia di seconda linea: agenti immunosoppressori
- Terapia di terza linea: farmaci biologici
- Prognosi e monitoraggio
- Gestione delle complicanze croniche

Caratterizzazione istopatologica
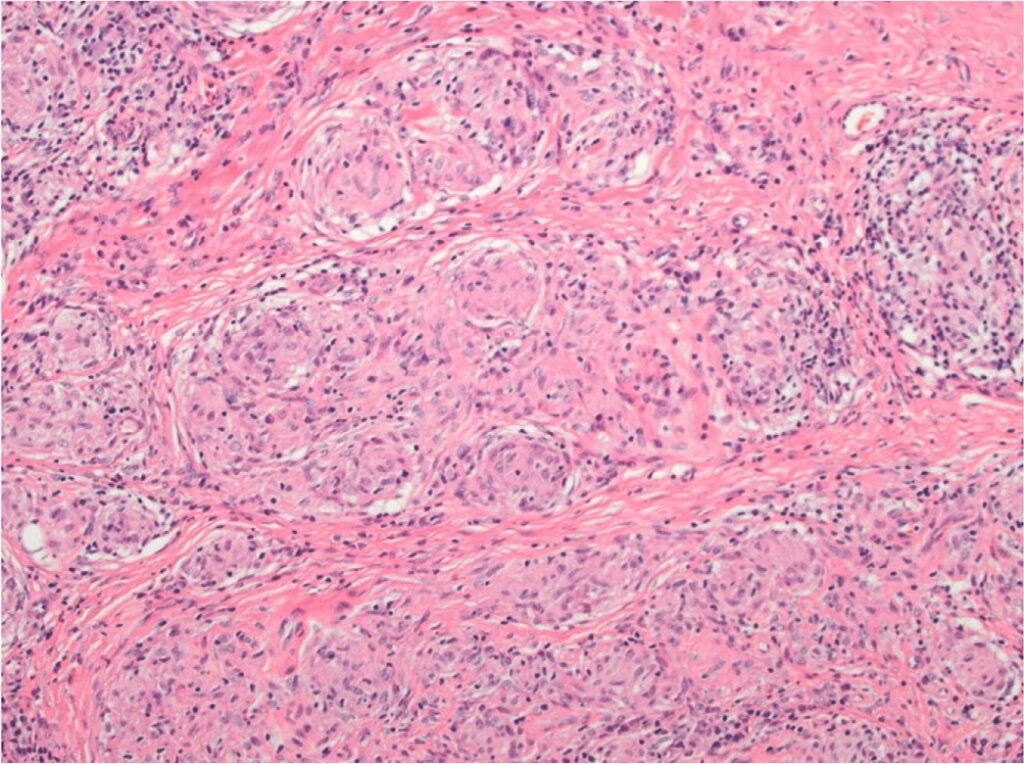
Dal punto di vista istopatologico, il granuloma sarcoideo rappresenta l’unità funzionale della malattia. Esso è costituito da un raggruppamento organizzato di cellule infiammatorie, principalmente macrofagi attivati che assumono un aspetto epitelioide e possono fondersi per formare cellule giganti multinucleate, come le cellule di Langhans.4
Questi aggregati sono tipicamente circondati da una corona di linfociti, con una netta prevalenza di linfociti T helper CD4+, e in misura minore da linfociti B e plasmacellule.5 Un elemento discriminante fondamentale rispetto alla tubercolosi è l’assenza di necrosi caseosa centrale, sebbene in rari casi possano essere presenti minime aree di necrosi fibrinoide.
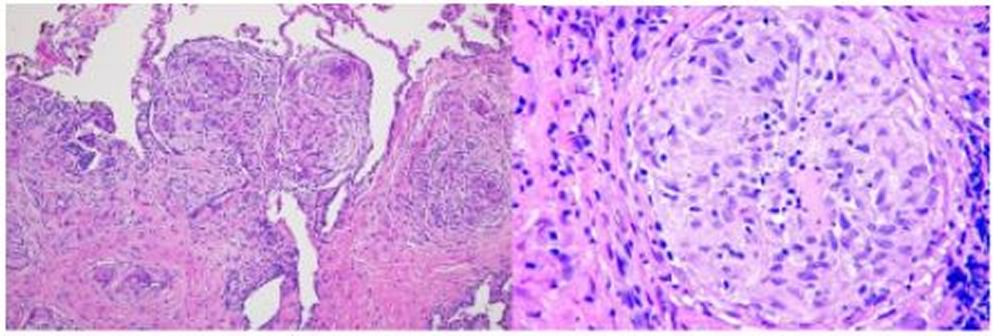
A sinistra (basso ingrandimento): presenza di granulomi distribuiti lungo il fascio broncovascolare; a destra (alto ingrandimento): dettaglio di un granuloma con cellule giganti multinucleate al centro.
Colorazione con ematossilina ed eosina, ingrandimento 400x. Granulomi non caseificanti nel derma con istiociti multinucleati e infiammazione linfocitaria.
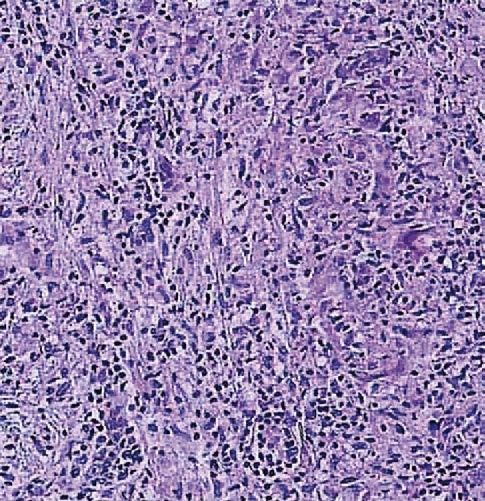
Gli stessi reperti istopatologici possono essere osservati in un’elevata percentuale di pazienti con patologie sarcoidi-simili.
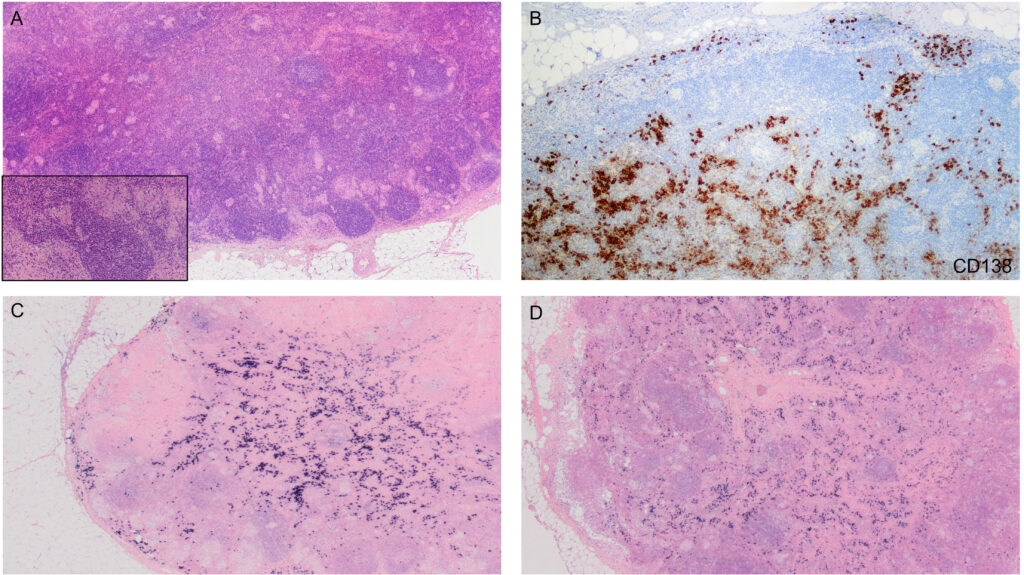
(B) CD138: proliferazione plasmacellulare positiva.
(C, D) Catene leggere: assenza di restrizione per le catene kappa o lambda.
Caricamento....
La dinamica di formazione del granuloma suggerisce un processo di compartimentazione immunitaria, in cui l’organismo tenta di isolare un antigene persistente e non degradabile. Se il processo infiammatorio non si risolve, la persistenza delle citochine profibrotiche prodotte dai macrofagi e dai linfociti Th2 può indurre una transizione verso la fibrosi, portando alla distruzione dell’architettura tissutale originale e alla perdita della funzione d’organo, come osservato nellostadio IV della sarcoidosi polmonare.6 7

Architettura epidemiologica e varianti demografiche
L’epidemiologia della sarcoidosi riflette una complessa interazione tra fattori ereditari, esposizioni ambientali e disparità socio-economiche, manifestandosi con variazioni geografiche, etniche e di genere estremamente marcate.
A livello globale, la prevalenza è stimata tra 1 e 5 casi ogni 10.000 abitanti, ma questa statistica generale maschera differenze locali profonde. L’incidenza annuale oscilla tra 2 e 40 casi per 100.000 persone, con i tassi più elevati registrati nei paesi del Nord Europa e tra la popolazione afroamericana negli Stati Uniti.
| Parametro epidemiologico | Dettaglio clinico e demografico |
|---|---|
| Prevalenza globale | 1-5 per 10.000 abitanti |
| Incidenza in Europa | 1-625 per 500.000 abitanti (estrema variabilità) |
| Picco di età (primo) | 20-45 anni (prevalenza tra la terza e quinta decade) |
| Picco di età (secondo) | Donne di età superiore ai 50 anni |
| Rapporto di genere | Lieve predilezione femminile (fino a 2:1 in alcune coorti) |
| Etnia afroamericana | Incidenza di 35,5 per 100.000; forme più gravi e croniche |
| Popolazione scandinava | Alta prevalenza; frequente Sindrome di Löfgren (1/3 dei casi) |
In Italia, la sarcoidosi è ufficialmente riconosciuta come malattia rara. I dati provenienti da registri regionali, come quelli del Piemonte e della Valle d’Aosta, indicano una prevalenza di circa 19,5 casi su 100.000 persone.8 Altre stime nazionali suggeriscono un tasso di circa 1 individuo ogni 10.000 abitanti.
L’analisi dei flussi migratori e dei cambiamenti demografici indica che la sarcoidosi sta diventando una diagnosi sempre più frequente anche in popolazioni precedentemente considerate a basso rischio, probabilmente a causa di una maggiore consapevolezza clinica e di una migliore accessibilità alle tecniche di imaging avanzate.
Caricamento…
Eziopatogenesi
L’eziologia della sarcoidosi rimane l’aspetto più dibattuto della patologia. L’ipotesi corrente la configura come una malattia multifattoriale, derivante dall’esposizione di un ospite geneticamente suscettibile a uno o più agenti scatenanti di natura infettiva o non infettiva.
Suscettibilità genetica
La componente genetica è supportata da studi su gemelli e dall’osservazione di cluster familiari, che rappresentano circa il 5-10% dei casi.9
La ricerca si è concentrata sul sistema dell’antigene leucocitario umano (HLA), identificando specifici alleli che influenzano non solo il rischio di contrarre la malattia, ma anche il suo decorso clinico e la prognosi. Ad esempio, l’allele HLA–DRB1∗03 è strettamente associato alla sindrome di Löfgren, una forma acuta a risoluzione spontanea, mentre alleli come HLA–DRB1∗11, ∗12, ∗14 e ∗15 sono correlati a una maggiore suscettibilità generale e, in alcuni casi, a un fenotipo cronico o fibrosante.
Trigger ambientali e infettivi
Tra i potenziali agenti scatenanti ambientali, sono stati identificati polveri di metalli (come il berillio, che causa una patologia quasi indistinguibile, la berilliosi), insetticidi, muffe, solventi e resine vegetali.
Anche il personale sanitario sembra presentare un rischio relativo aumentato, suggerendo una possibile trasmissione di agenti infettivi non ancora isolati o una specifica esposizione professionale.
L’ipotesi infettiva punta fortemente sui micobatteri (in particolare varianti simili a Mycobacterium tuberculosis) e sul Propionibacterium acnes, le cui componenti proteiche sono state rinvenute all’interno dei granulomi di pazienti sarcoidei.
Meccanismi immunologici della granulomatosi
Il processo inizia quando un antigene sconosciuto viene fagocitato dalle cellule presentanti l’antigene (APC), come imacrofagi alveolari e le cellule dendritiche. Queste cellule migrano verso i linfonodi drenanti, secernendo citochine come IL-12 e IL-1, che promuovono l’espansione clonale dei linfociti T helper di tipo 1 (Th1) e Th17.10 Questi linfociti producono elevate quantità di interferone-gamma (IFN–γ) e interleuchina-2 (IL-2), che a loro volta attivano ulteriormente i macrofagi, stimolando la produzione di fattore di necrosi tumorale alfa (TNF–α).
Il TNF–α agisce come una molecola chiave per il reclutamento cellulare e l’organizzazione architettonica del granuloma.
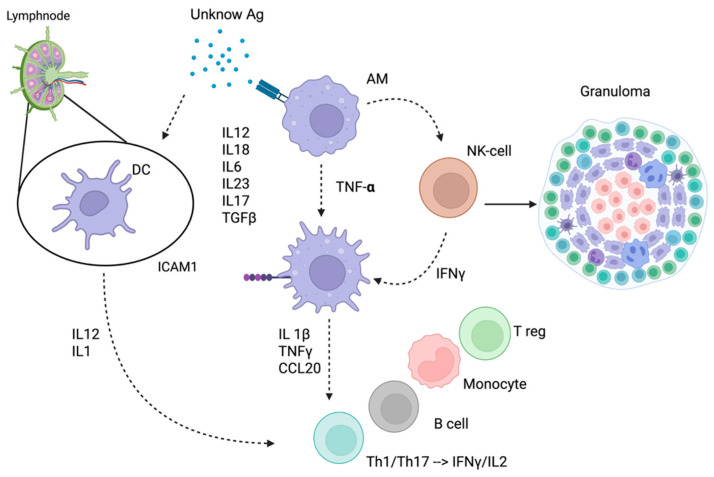
Un antigene ignoto (Ag) innesca l’attivazione dei macrofagi alveolari (AM) e delle cellule dendritiche (DC). Le DC migrano ai linfonodi drenanti rilasciando IL-12 e IL-1, citochine che guidano la differenziazione e l’espansione dei linfociti Th1 e Th17 (produttori di IFN-γ e IL-2). Il TNF-α stimola le cellule NK a rilasciare IFN-γ, il quale amplifica ulteriormente l’attivazione macrofagica. I macrofagi attivati up-regolano molecole di adesione (es. ICAM-1) e secernono un ampio pannello di mediatori pro-infiammatori (tra cui IL-1β, TNF-α, IL-17 e TGF-β), creando un microambiente che recluta cellule immuni (linfociti B, T e monociti) e sostiene l’architettura strutturale del granuloma.
Reazioni sarcoidee indotte da farmaci e biotecnologie (DISR)
Un’area di estrema rilevanza nella pratica clinica moderna è quella delle reazioni sarcoidee indotte da farmaci (Drug-Induced Sarcoid-like Reactions, DISR), manifestazioni granulomatose sistemiche che mimano fedelmente la sarcoidosi idiopatica ma insorgono in correlazione temporale con l’assunzione di specifiche terapie.
Queste reazioni rappresentano un paradosso immunologico, specialmente quando scatenate da farmaci nati per sopprimere l’infiammazione o per trattare la sarcoidosi stessa.
Inibitori dei Checkpoint Immunitari (ICI)

Gli ICI (nivolumab, pembrolizumab, ipilimumab, avelumab) hanno trasformato la terapia oncologica sbloccando i meccanismi di evasione immunitaria dei tumori. Tuttavia, rimuovendo i freni inibitori dei linfociti T, possono scatenare eventi avversi immuno-correlati (irAE), tra cui le DISR.11
Il blocco di CTLA-4 eleva i livelli circolanti di cellule Th17, aumentando la produzione di IL-17 e TNF–α, mentre il blocco di PD-1/PD–L1 potenzia l’attività dei Th17 e può attivare la via metabolica di mTOR, essenziale per la sopravvivenza dei macrofagi nel granuloma. Clinicamente, queste reazioni possono essere confuse con la progressione del cancro a causa della comparsa di nuove linfoadenopatie ilari o mediastiniche captanti alla PET.12
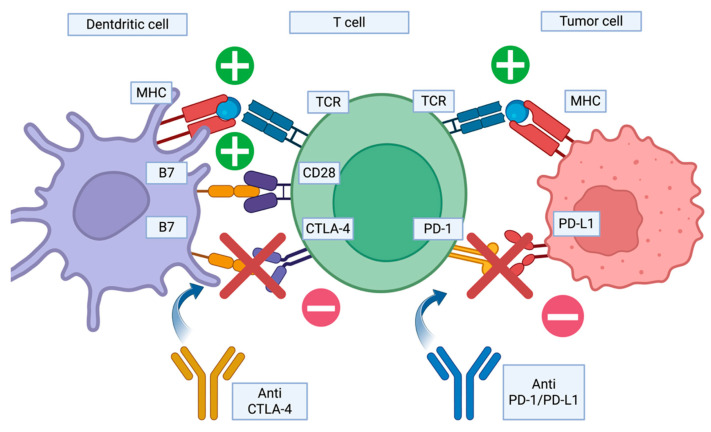
Gli ICI agiscono bloccando i pathway inibitori che sopprimono la risposta immunitaria. Ipilimumab ha come bersaglio il CTLA-4, espresso sui linfociti T attivati a livello degli organi linfoidi. A livello dei tessuti periferici, Nivolumab e Pembrolizumab bloccano il recettore PD-1 sui linfociti T, mentre Atezolizumab, Durvalumab e Avelumab inibiscono il ligando PD-L1, espresso sia sulle cellule tumorali che sulle cellule immunitarie
Antagonisti del TNF-alfa
Sebbene farmaci come infliximab e adalimumab siano usati come terza linea nel trattamento della sarcoidosi refrattaria, essi possono indurre reazioni sarcoidee. Questo fenomeno sembra legato a un’alterazione dell’equilibrio tra i recettori del TNF. Il blocco del TNF può interferire con la via del recettore 2 (TNFR2), che è cruciale per la funzione soppressoria delle cellule T regolatorie (Treg).
Una riduzione dell’attività delle Treg può portare a un rimbalzo della risposta Th1/Th17, facilitando la formazione di granulomi. L’etanercept, agendo come un recettore solubile, sembra essere associato più frequentemente a questo effetto rispetto agli anticorpi monoclonali completi.
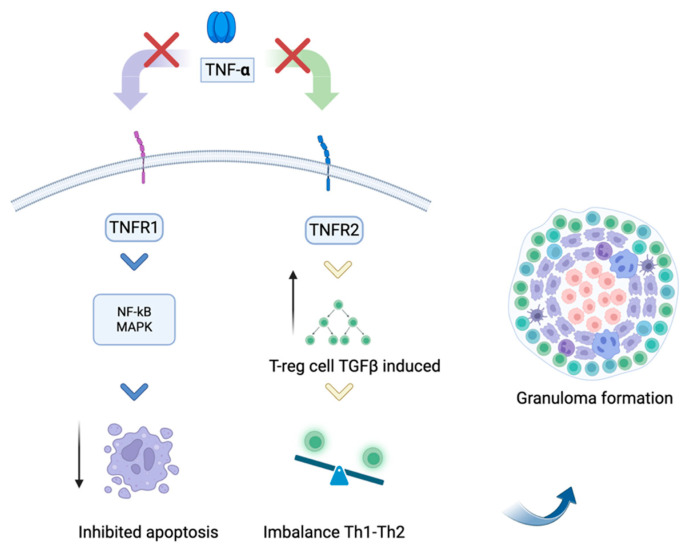
Gli agenti anti-TNF legano selettivamente il TNF-α, inibendone l’interazione con il recettore TNFR1 (p55); ciò previene l’attivazione di NF-kB, delle MAPK e delle vie apoptotiche. Parallelamente, il blocco del legame con TNFR2 (p75) induce un significativo incremento delle cellule Treg che, attraverso la produzione di TGF-β, contribuiscono alla regolazione immunitaria e alla dinamica di formazione del granuloma.
Caricamento....
| Categoria farmacologica | Esempi principali | Meccanismo ipotizzato della DISR |
|---|---|---|
| Inibitori Checkpoint (ICI) | Pembrolizumab, Nivolumab, Ipilimumab | Iperattivazione T-cellulare; aumento Th17; attivazione via mTOR |
| Antagonisti TNF-α | Etanercept, Adalimumab, Infliximab | Squilibrio TNFR1/TNFR2; inibizione cellule Treg; effetto paradossale |
| Inibitori di BRAF | Vemurafenib | Alterazione profili citochinici; attivazione MAPK paradossale |
| Interferoni | IFN-alfa, IFN-beta | Stimolazione diretta della risposta Th1 e attivazione macrofagica |
| Terapia antiretrovirale | HAART | Sindrome da ricostituzione immunitaria (IRIS) |

Clinica
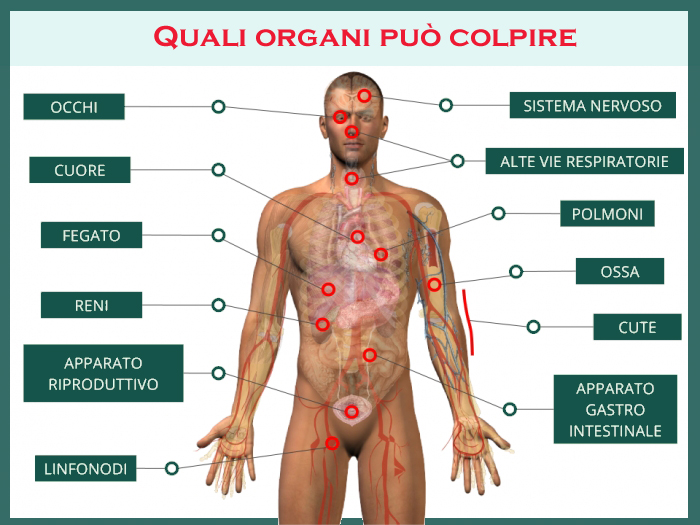
La sarcoidosi è definita come “la grande imitatrice” per la sua capacità di interessare quasi ogni organo, manifestandosi con sintomi che variano drasticamente in base alla sede e all’estensione del coinvolgimento granulomatoso.
Circa un terzo dei pazienti presenta sintomi sistemici prodromici, tra cui astenia (che può essere invalidante e non correlata all’attività della malattia), febbricola, calo ponderale e sudorazioni notturne.
Apparato respiratorio (>90%)
La sarcoidosi polmonare si presenta tipicamente con tosse secca e stizzosa, dispnea da sforzo che può progredire verso la dispnea a riposo nelle forme avanzate, e dolore toracico aspecifico.
All’esame obiettivo, il torace può risultare sorprendentemente silente, anche in presenza di estese alterazioni radiografiche, sebbene in alcuni casi siano udibili fini crepitii basali.
L’emottisi è estremamente rara e deve far sospettare complicanze come il micetoma in cavità fibrotiche preesistenti.
Manifestazioni cutanee (25%)
La pelle rappresenta una “finestra” diagnostica fondamentale. Le lesioni si dividono in specifiche (contenenti granulomi) e aspecifiche:
- eritema nodoso: manifestazione aspecifica caratterizzata da noduli rossi, caldi e dolenti sulle superfici estensorie delle gambe. È spesso associato a una prognosi favorevole e fa parte della sindrome di Löfgren;
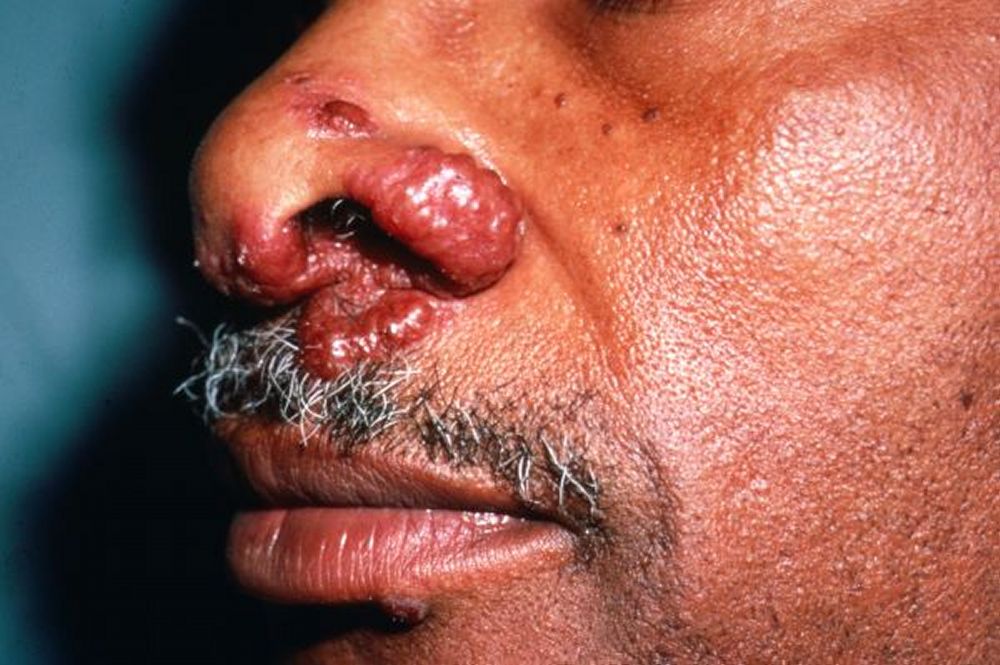
- Lupus Pernio: placche rosso-violacee indurite localizzate sul naso, guance e orecchie. È un segno di malattia cronica, spesso associato a coinvolgimento delle vie respiratorie superiori e a una risposta terapeutica meno brillante;
- lesioni cicatriziali: i granulomi possono infiltrarsi in vecchie cicatrici o tatuaggi, causandone il rigonfiamento e l’arrossamento, un segno clinico molto suggestivo.
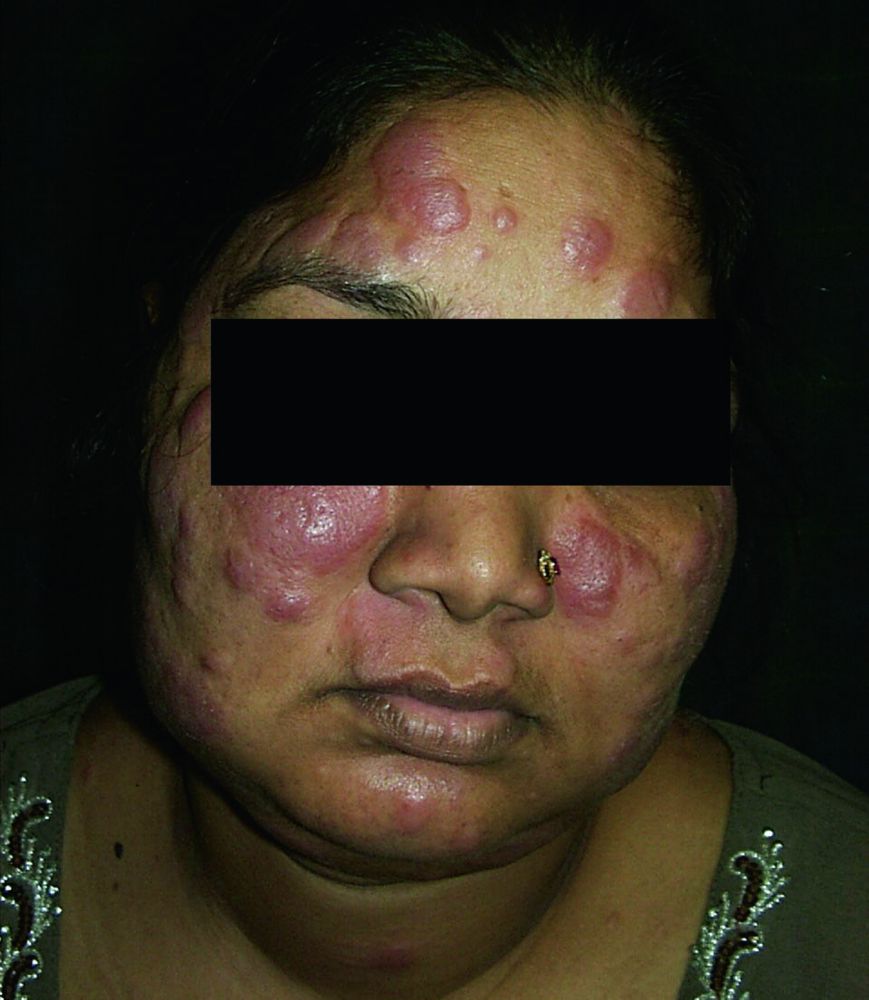
Questa foto mostra placche facciali eritematose in un paziente con sarcoidosi.
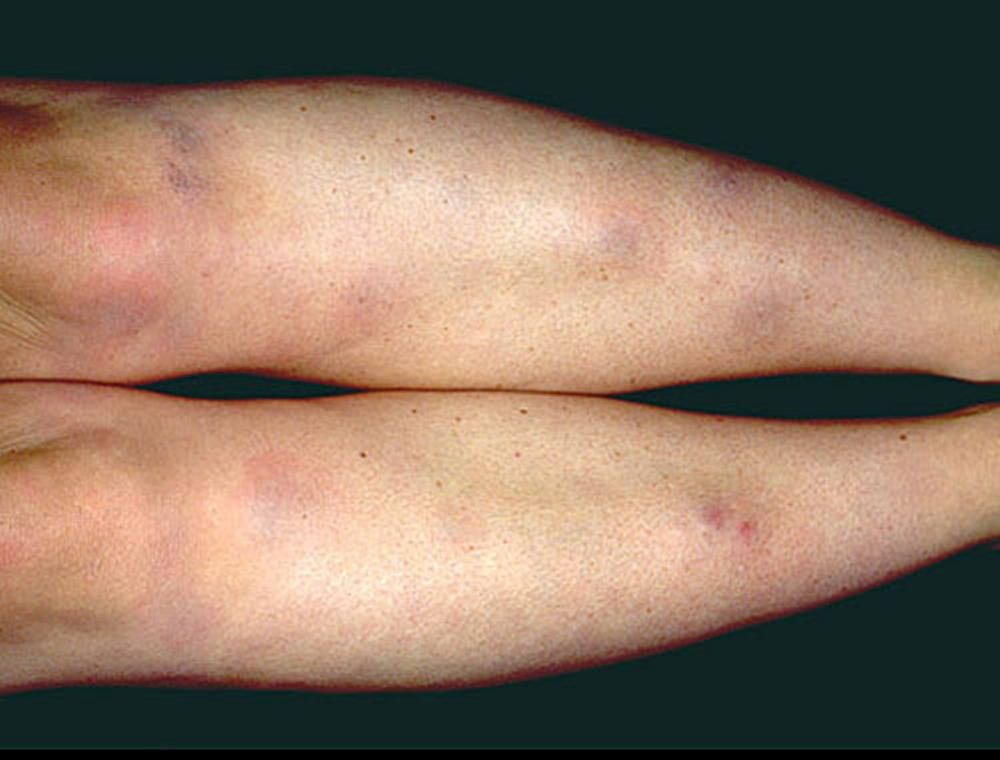
Questa foto mostra i caratteristici noduli sottocutanei dolenti, eritematosi, sugli stinchi di un paziente con eritema nodoso.
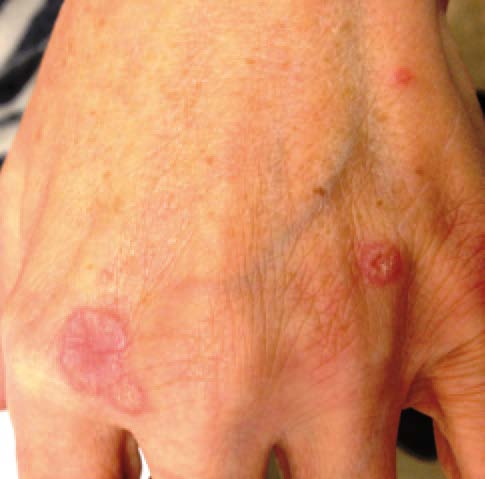
Questa immagine mostra noduli della pelle in un paziente con sarcoidosi.
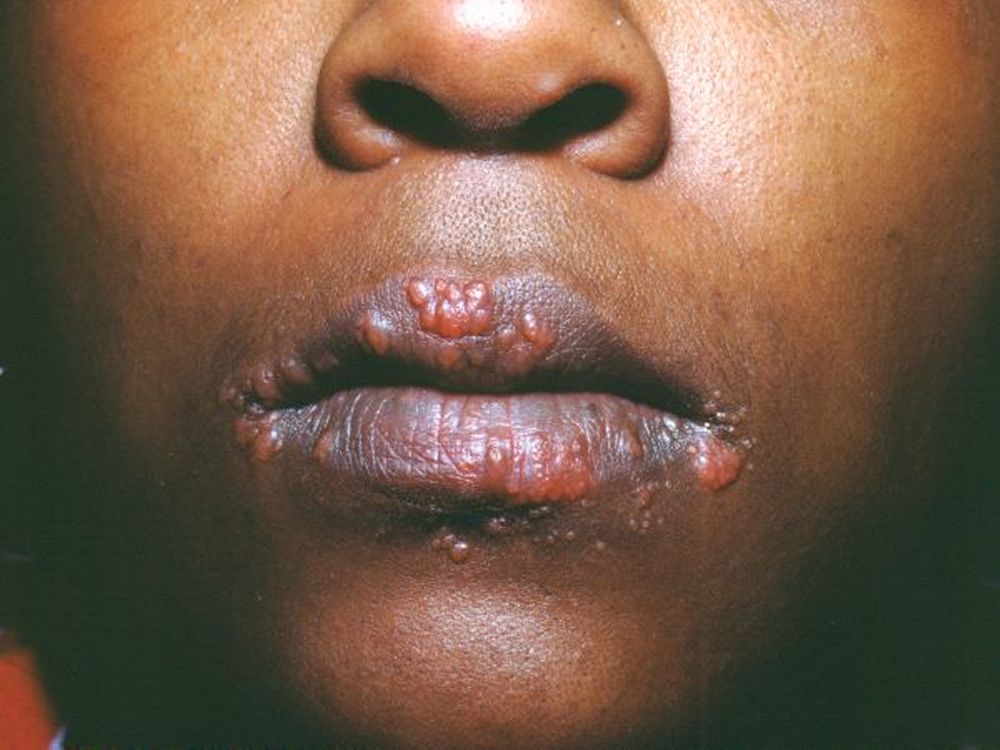
Questa immagine mostra papule multiple sulle labbra di un paziente con sarcoidosi.
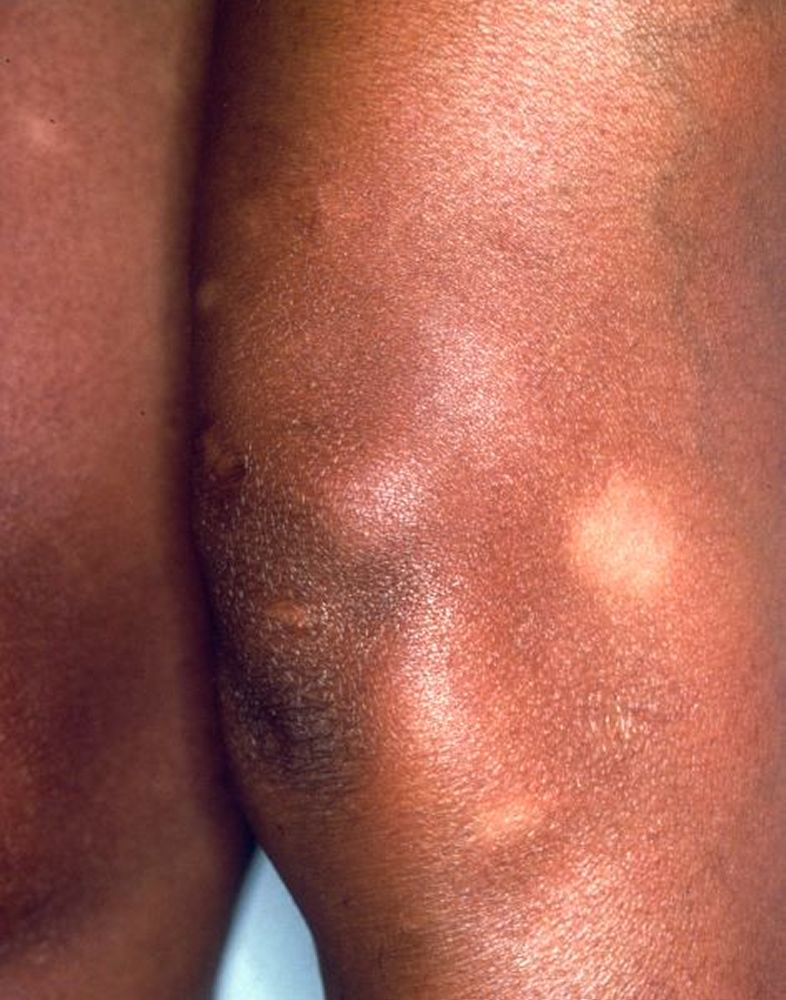
Aree ipopigmentate e noduli sottocutanei in un paziente con sarcoidosi.
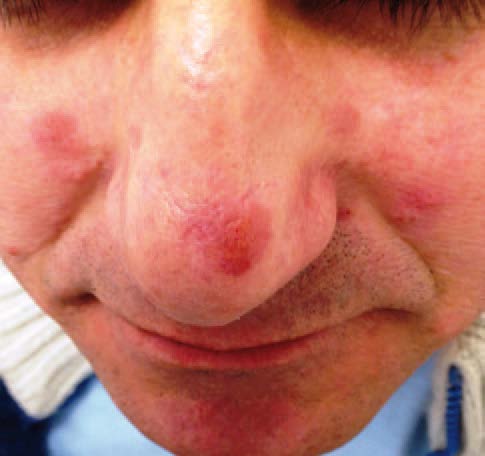
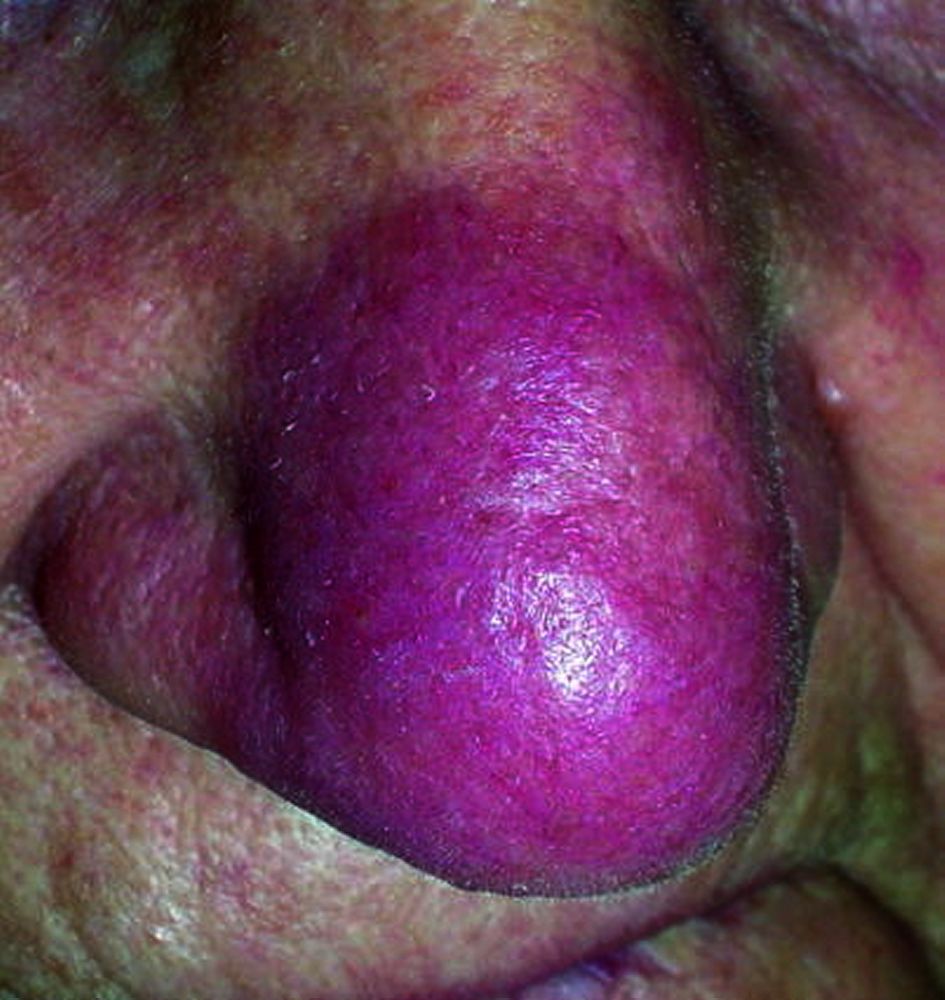
Questa foto mostra placche violacee sul naso di un paziente con sarcoidosi.
Coinvolgimento oculare (12-25%)
L’occhio è interessato in una percentuale significativa di pazienti, con variazioni etniche (più comune negli afroamericani e giapponesi).
L’uveite anteriore è la forma più frequente, manifestandosi con occhio rosso, dolore e fotofobia.
L’uveite cronica o posteriore può essere asintomatica nelle fasi iniziali ma può portare a cataratta, glaucoma e cecità se non identificata tramite screening periodici con lampada a fessura.
Sarcoidosi cardiaca e neurologica
Caricamento….
Queste localizzazioni, sebbene meno comuni, sono responsabili della maggior parte della mortalità e morbilità grave:
- cuore: il coinvolgimento clinico è raro (<5%), ma gli studi autoptici suggeriscono una prevalenza molto più alta (25%). Si manifesta con disturbi della conduzione (blocco atrioventricolare di grado elevato), aritmie ventricolari e scompenso cardiaco da cardiomiopatia restrittiva;13
- sistema nervoso (<10%): la neurosarcoidosi può colpire il sistema nervoso centrale o periferico. La paralisi del nervo facciale (VII) è il segno più comune, spesso bilaterale o alternante. Altre manifestazioni includono meningite basale, diabete insipido per coinvolgimento ipofisario e neuropatie periferiche.
Percorso diagnostico e stratificazione del rischio
La diagnosi di sarcoidosi non si fonda su un singolo test patognomonico, ma sulla convergenza di evidenze cliniche, radiologiche e istologiche, previa esclusione di altre condizioni granulomatose.14
Stadiazione radiografica di Scadding
La radiografia del torace (RX) rimane il punto di partenza per la classificazione della sarcoidosi polmonare, utilizzando il sistema di Scadding che divide la malattia in quattro stadi principali sulla base dei reperti linfo-parenchimali.[1, 12, 13]
| Stadio | Definizione radiografica | Significato clinico e prognostico |
|---|---|---|
| 0 | RX torace normale | Malattia puramente extratoracica o pre-radiologica |
| I | Linfoadenopatia ilare bilaterale (BHL) | Stadio più comune; spesso asintomatico; alta remissione spontanea |
| II | BHL associata a infiltrati parenchimali | Presenza di granulomi sia nei linfonodi che nel tessuto polmonare |
| III | Infiltrati parenchimali senza BHL | Granulomi confinati al polmone; minore probabilità di remissione |
| IV | Fibrosi polmonare diffusa | Presenza di cicatrici permanenti, perdita di volume e cisti |
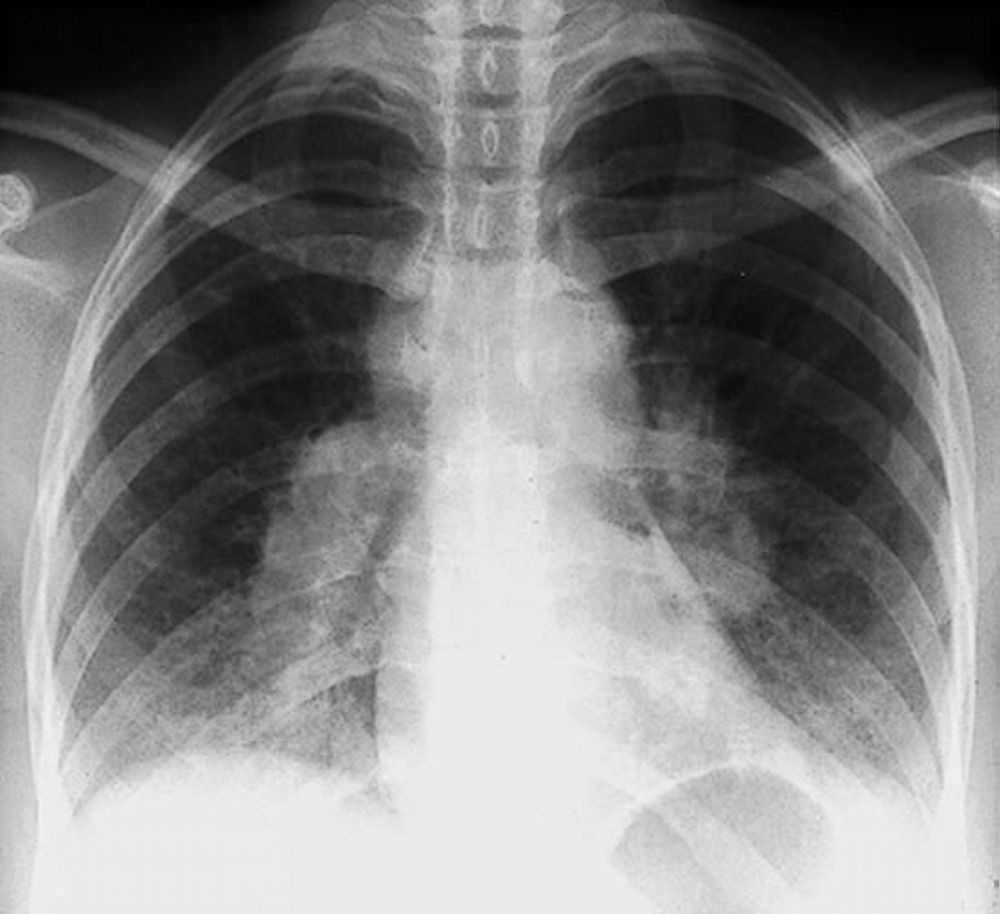
Adenopatia ilare bilaterale nella sarcoidosi di stadio I.
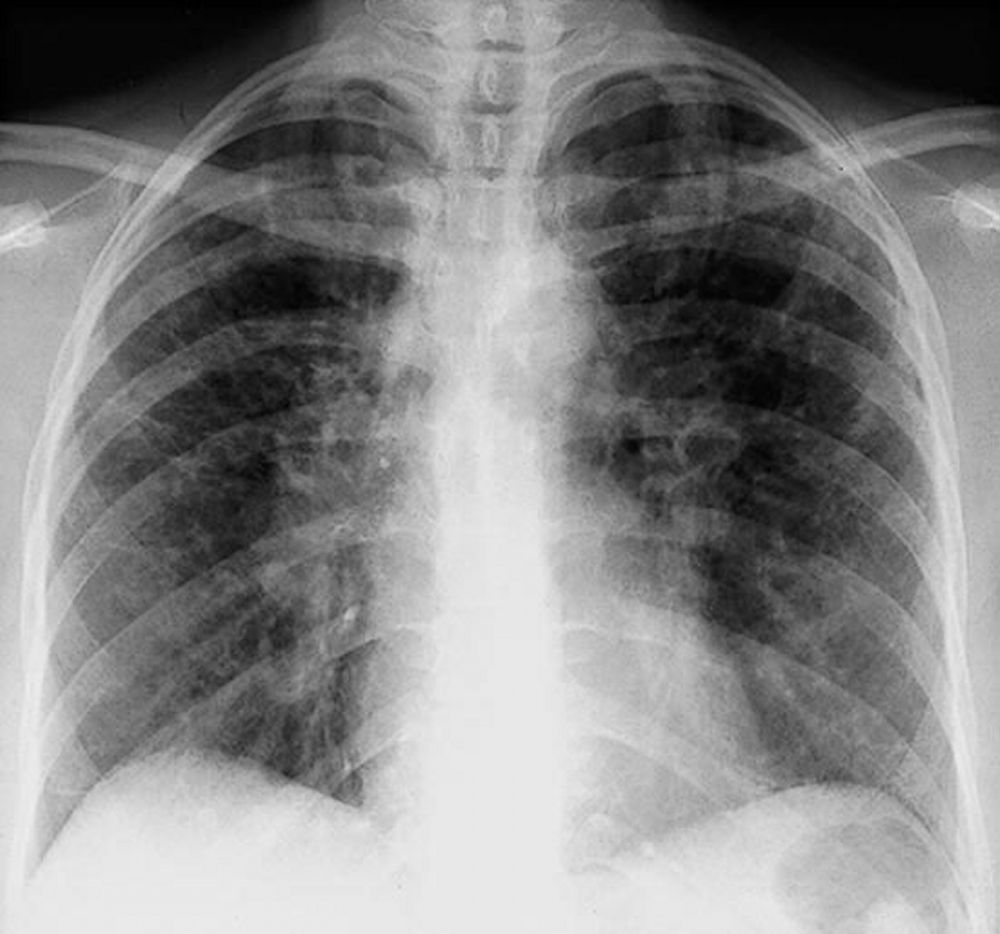
Adenopatie ilari bilaterali con opacità interstiziali nella sarcoidosi al II stadio.
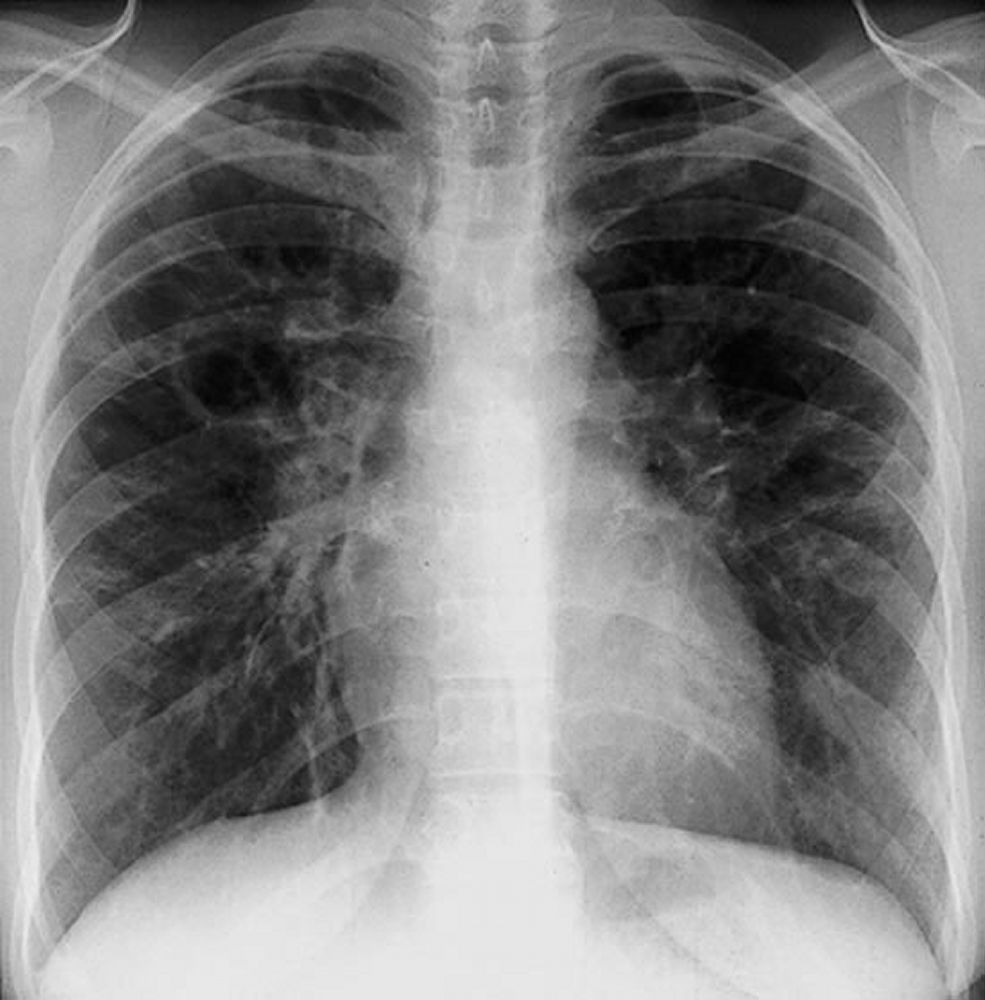
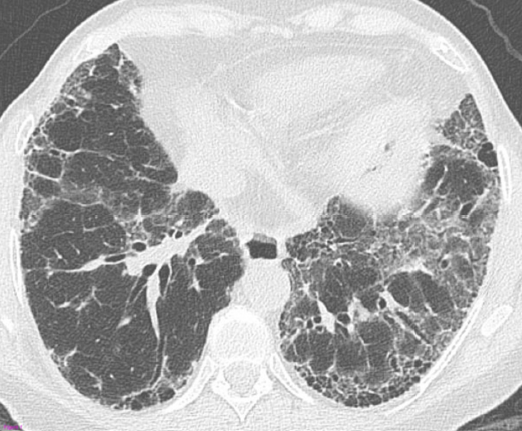
Opacità interstiziali diffuse senza adenopatia ilare nello stadio III della sarcoidosi.
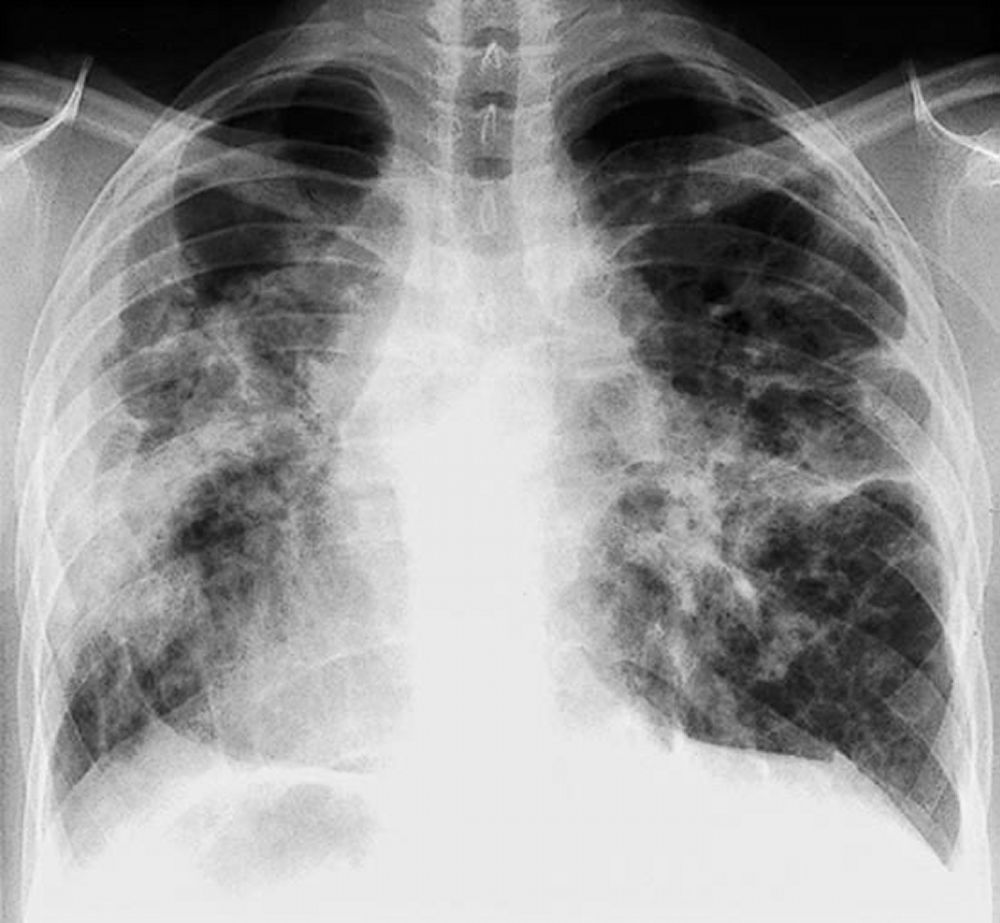
Grave fibrosi diffusa con adenopatia ilare e alterazioni cistiche dei lobi superiori nella sarcoidosi allo stadio IV.
Tuttavia, la TC ad alta risoluzione (HRCT) è oggi considerata indispensabile per una migliore caratterizzazione. I segni tipici alla HRCT includono noduli a distribuzione perilinfatica (lungo i setti interlobulari e le scissure), ispessimento dei fasci broncovascolari e opacità a “vetro smerigliato” che riflettono un’alveolite granulomatosa attiva.
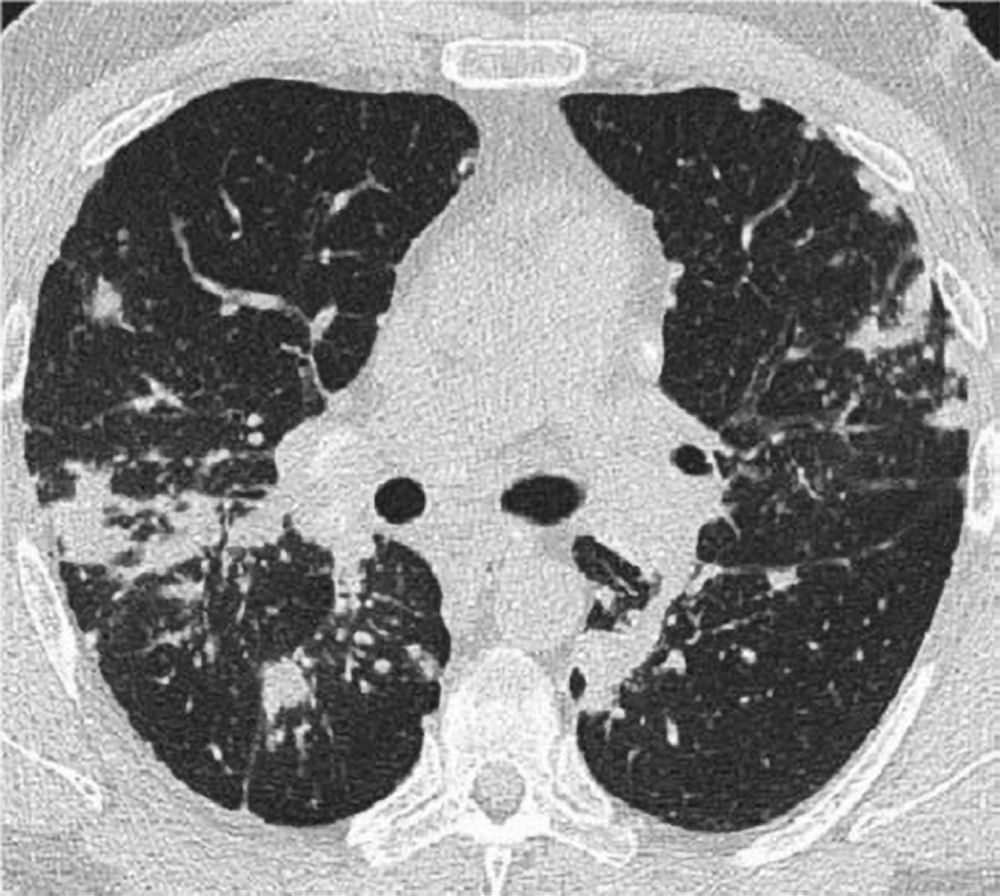
che mostra ispessimento dei fasci broncovascolari e perline dei setti interlobulari.
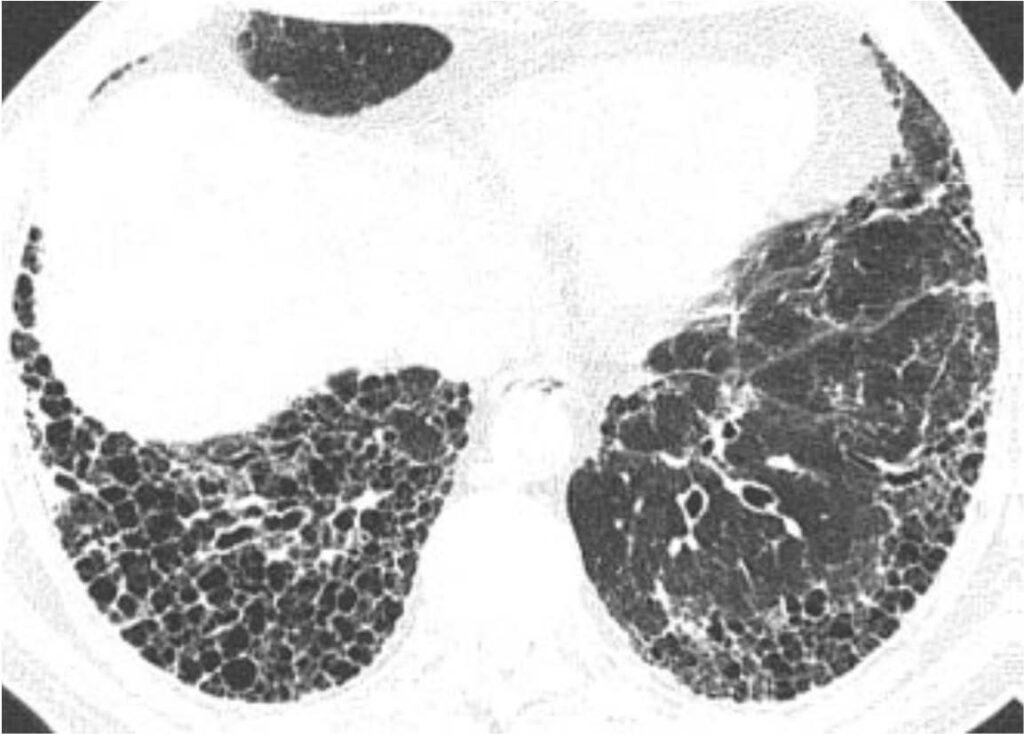
nel polmone di sinistra), bronchiectasie da trazione, opacità reticolari e aree di ground glass
Caricamento....
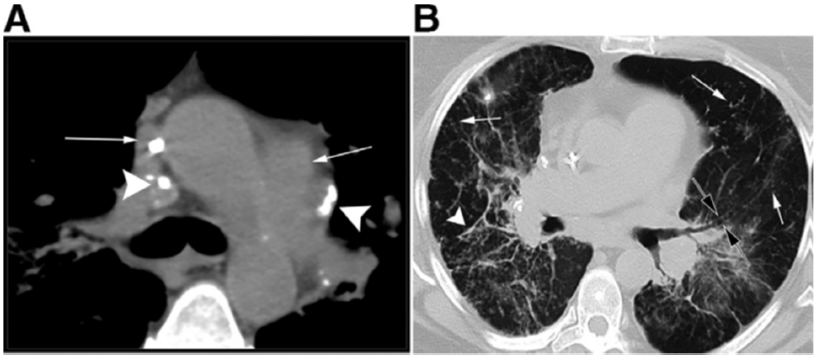
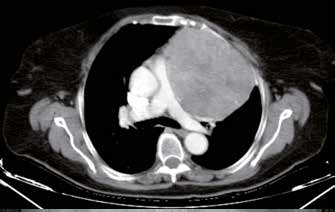
(A) TC coronale senza mezzo di contrasto: evidenti linfoadenopatie mediastiniche simmetriche (frecce bianche) con calcificazioni (punta di freccia).
(B) Parenchima polmonare: opacità reticolari (punte di freccia bianche), estese bronchiectasie da trazione (frecce nere) e noduli perilinfatici (frecce bianche). Quadro suggestivo di sarcoidosi polmonare.

PET-TC con FDG
La Tomografia a Emissione di Positroni con FDG rappresenta uno strumento avanzato e fondamentale nella gestione della sarcoidosi, in particolare per i casi complessi o quando le tecniche standard (come la RX o la TC) non forniscono risposte definitive.
La FDG-PET non si limita a mostrare l’anatomia (come la TC), ma evidenzia l’attività metabolica della malattia. I granulomi attivi captano il glucosio marcato, apparendo come aree “luminose” o “calde” nelle immagini. Le sue funzioni principali sono:

- valutazione dell’attività infiammatoria: è in grado di distinguere tra infiammazione attiva (che richiede terapia) e lesioni cicatriziali o fibrotiche (che non rispondono agli antinfiammatori);
- stadiazione sistemica: essendo un esame total body, permette di individuare localizzazioni occulte della malattia in organi non sospetti, specie nel cuore nel sospetto clinico di sarcoidosi cardiaca anche se l’ECG e l’ecocardiogramma risultano negativi;
- guida alla biopsia: grazie alla sua capacità di individuare le aree a maggior attività metabolica, aiuta il chirurgo o lo pneumologo a scegliere il sito migliore per effettuare il prelievo, aumentando le probabilità di trovare granulomi;
- risposta terapeutica: la riduzione della captazione (SUVmax) alla PET dopo il trattamento (es. con Infliximab) è correlata al miglioramento della funzionalità polmonare (FVC), suggerendo che la PET sia utile per decidere se continuare o modificare le terapie in casi complessi.
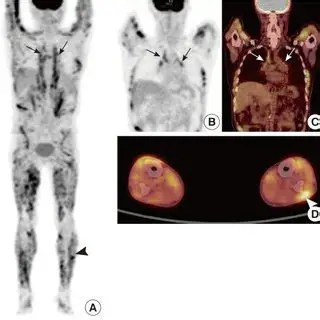
Nonostante i suoi vantaggi, la PET presenta delle criticità:
- non specificità: l’aumentata captazione di FDG non è esclusiva della sarcoidosi; si verifica anche in infezioni e tumori. Ad esempio, sia i linfonodi sarcoidei che le metastasi tumorali captano il glucosio, rendendo talvolta difficile la distinzione senza una biopsia;
- reazioni sarcoidee indotte da farmaci (DISR): nei pazienti oncologici trattati con inibitori dei checkpoint immunitari (es. nivolumab, ipilimumab), la comparsa di linfonodi captanti alla PET può simulare una progressione del tumore, mentre si tratta in realtà di una reazione simil-sarcoidea.

Biopsia e tecniche endoscopiche
L’evidenza istologica di granulomi non caseosi è necessaria nella maggior parte dei casi per confermare la diagnosi. La scelta del sito bioptico deve ricadere sulle sedi più accessibili e meno invasive, come lesioni cutanee, linfonodi periferici o noduli congiuntivali. Se queste non sono disponibili, la fibrobroncoscopia è la procedura d’elezione:
- EBUS-TBNA (Endobronchial Ultrasound-guided Transbronchial Needle Aspiration): ha rivoluzionato la diagnostica, permettendo di campionare i linfonodi mediastinici e ilari con una sensibilità superiore al 90%, riducendo drasticamente la necessità di mediastinoscopia chirurgica;
- biopsia transbronchiale (TBB): utile per campionare il parenchima polmonare se l’EBUS non è dirimente o se si sospettano patologie parenchimali alternative.
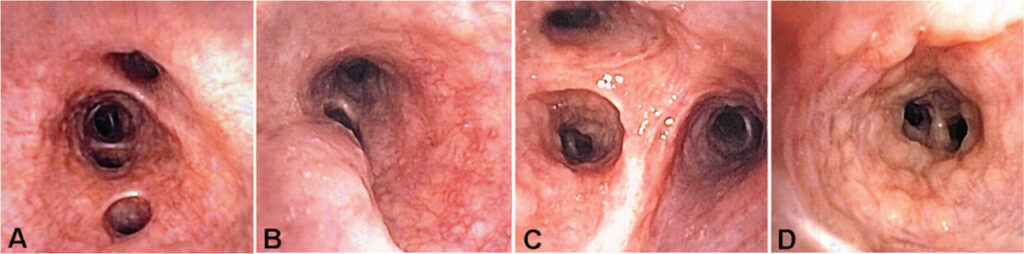
(A) Bronco intermedio: eritema, edema della mucosa e aspetto granulomatoso.
(B) Ipervascolarizzazione capillare, noduli multipli ed eritema.
(C) Alterazioni analoghe nel bronco segmentale del lobo superiore destro.
(D) Tipici noduli mucosi (3-4 mm) in un bronco segmentale.
Laboratorio e altre indagini
Il dosaggio dell’ACE sierico è storicamente associato alla sarcoidosi; tuttavia, la sua utilità è limitata dalla bassa sensibilità e specificità. Livelli elevati si riscontrano solo nel 40-60% dei pazienti con malattia attiva. Più rilevante è il monitoraggio del metabolismo del calcio (calcemia e calciuria delle 24 ore) e della funzionalità renale, dato il rischio di ipercalcemia causata dalla produzione endogena di vitamina D da parte dei macrofagi granulomatosi.
La spirometria completa di DLCO (capacità di diffusione del monossido di carbonio) permette di valutare il deficit ventilatorio, tipicamente di tipo restrittivo, e l’impatto funzionale dell’inspessimento interstiziale.
Caricamento…
Diagnosi differenziale e identificazione dei “Mimics”
La diagnosi differenziale è una fase critica per evitare errori terapeutici potenzialmente catastrofici, specialmente l’uso di steroidi in presenza di infezioni misconosciute:
Caricamento…
- infezioni granulomatose: la tubercolosi (TB) e le micosi profonde (come l’istoplasmosi) devono essere escluse tramite esami colturali, PCR e test cutanei/ematici (IGRA), specialmente in pazienti provenienti da aree endemiche o con sintomi sistemici marcati;
- pneumopatie professionali: la berilliosi cronica è clinicamente e istologicamente identica alla sarcoidosi; la diagnosi dipende da un’anamnesi lavorativa accurata (settori aerospaziale, elettronica, metallurgia) e dal test di proliferazione linfocitaria al berillio (BeLPT);
- neoplasie e reazioni paraneoplastiche: i linfomi possono presentarsi con linfoadenopatie ilari bilaterali captanti alla PET. Inoltre, alcuni tumori solidi possono scatenare “reazioni simil-sarcoidee” nei linfonodi drenanti, che non indicano necessariamente una sarcoidosi sistemica;
- altre malattie sistemiche: la granulomatosi con poliangite (GPA), la polmonite da ipersensibilità (HP) e la malattia di Crohn possono presentare granulomi tissutali, ma solitamente con caratteristiche distintive (necrosi nella GPA, distribuzione bronchiolocentrica nella HP, sintomi gastrointestinali nella Crohn).
Caricamento….
Paradigmi terapeutici e Linee Guida Internazionali
Il trattamento della sarcoidosi è guidato dal principio del bilanciamento tra il rischio di progressione della malattia e la tossicità dei farmaci.15 Poiché la remissione spontanea è comune, molti pazienti (soprattutto in stadio I o II asintomatici) richiedono solo un monitoraggio clinico-radiologico stretto (“wait and watch“).
Terapia di prima linea: glucocorticoidi
I corticosteroidi sistemici (prednisone/prednisolone) rappresentano il trattamento iniziale per la malattia sintomatica o con minaccia di danno d’organo. Le linee guida ERS suggeriscono una dose iniziale moderata di 20 mg/die (o 0,5 mg/kg), evitando i regimi ad alte dosi che non hanno dimostrato superiorità ma aumentano significativamente gli effetti collaterali (diabete, ipertensione, osteoporosi, obesità).
Il trattamento deve essere mantenuto per un periodo minimo di 6-12 mesi per prevenire recidive precoci.
Terapia di seconda linea: agenti immunosoppressori

L’aggiunta di farmaci “steroid-sparing” è indicata quando la malattia non è controllata da dosi accettabili di steroidi (<10 mg/die) o quando i pazienti sviluppano tossicità intollerabile:
- methotrexate: è l’agente di seconda linea più comune. Dosato a 10-15 mg una volta a settimana, è efficace nel migliorare la funzione polmonare e nel permettere la riduzione del prednisone;
- azatioprina e leflunomide: rappresentano valide alternative nei pazienti che non tollerano il methotrexate, sebbene richiedano un monitoraggio attento dell’emocromo e della funzione epatica;
- idrossiclorochina: particolarmente efficace per le manifestazioni cutanee e per il controllo dell’ipercalcemia/ipercalciuria.
Terapia di terza linea: farmaci biologici
Nei casi di sarcoidosi refrattaria o multiorgano grave, si ricorre agli inibitori del TNF–α. L’infliximab è il biologico più studiato, con forti raccomandazioni per la neurosarcoidosi e la sarcoidosi oculare o cutanea grave.
L’adalimumab rappresenta una valida alternativa sottocutanea.
È mandatorio lo screening per la tubercolosi latente prima di iniziare questi farmaci.
| Farmaco | Dosaggio tipico | Principali tossicità | Monitoraggio raccomandato |
|---|---|---|---|
| Prednisone | Iniziale 20 mg/die; mantenimento 5-10 mg | Diabete, ipertensione, osteoporosi, cataratta | Glucosio, PA, densitometria ossea |
| Methotrexate | 10-15 mg/settimana | Nausea, leucopenia, epatotossicità | Emocromo, AST/ALT, Creatinina (ogni 1-3 mesi) |
| Infliximab | 3-5 mg/kg (settimane 0,2,6, poi ogni 4-8 settimane) | Infezioni gravi, reazioni allergiche, peggioramento CHF | Screening TB pre-trattamento; monitoraggio infezioni |
| Idrossiclorochina | 200-400 mg/die | Tossicità retinica (rara ma grave) | Visita oculistica periodica (6-12 mesi) |

Prognosi e monitoraggio
La prognosi della sarcoidosi è generalmente favorevole, ma una minoranza di pazienti affronta un decorso cronico e progressivo che impatta gravemente sulla qualità della vita.

Circa il 50-70% dei pazienti va incontro a remissione entro i primi 24 mesi dalla diagnosi. La recidiva è possibile in circa il 5% dei soggetti che hanno precedentemente raggiunto la remissione.
L’identificazione di fenotipi ad alto rischio è essenziale per un intervento precoce. I principali fattori associati a un decorso sfavorevole includono:
- esordio in età avanzata (>40 anni) ed etnia afroamericana;
- presenza di lupus pernio, coinvolgimento delle alte vie respiratorie o osseo;
- neurosarcoidosi, sarcoidosi cardiaca o uveite cronica;
- ipercalcemia cronica e nefrocalcinosi;
- presenza di fibrosi polmonare allo stadio IV di Scadding, ipertensione polmonare o severa riduzione della FVC e della DLCO.
Gestione delle complicanze croniche
L’affaticamento cronico (fatigue) colpisce fino all’ 80% dei pazienti e spesso persiste anche quando l’infiammazione granulomatosa è risolta. La gestione richiede programmi di riabilitazione polmonare e, in casi selezionati, l’uso di neurostimolanti come il metilfenidato o l’armodafinil.
Un’altra complicanza emergente è la sindrome delle apnee ostruttive del sonno (OSAS), che mostra una prevalenza aumentata nei pazienti sarcoidei e contribuisce significativamente alla sensazione di stanchezza diurna.
Il monitoraggio a lungo termine deve essere individualizzato, con visite ogni 3−6 mesi per i pazienti attivi e controlli annuali per quelli in remissione stabile.Gli esami di routine includono prove di funzionalità respiratoria, calcemia/calciuria, ECG e valutazione oculistica. La transizione dei pazienti pediatrici verso i centri dell’adulto deve essere gestita con percorsi strutturati per garantire la continuità delle cure.
Now loading…
FONTI:
- Sarcoidosi – Malattie polmonari – Manuali MSD Edizione Professionisti ↩︎
- Sarcoidosi – Orphanet ↩︎
- Sarcoidosis vs. Sarcoid-like reactions: The Two Sides of the same Coin? – ResearchGate ↩︎
- ↩︎
- Drug-Induced Sarcoid-like Reactions Associated to Targeted … ↩︎
- Cos’è la Sarcoidosi ↩︎
- Sarcoidosi: cos’è, come si manifesta e come si tratta – ISSalute ↩︎
- Sarcoidosi – Malattie Rare Piemonte ↩︎
- Sarcoidosi – European Lung Foundation ↩︎
- Drug-Induced Sarcoid-like Reactions Associated to Targeted Therapies and Biologic Agents ↩︎
- Case Report: The immune architecture of immunotherapy-induced cutaneous sarcoidosis resembles peritumoral inflammation – Frontiers ↩︎
- Hilar/mediastinal and cutaneous drug-induced sarcoidosis-like reaction associated with immune checkpoint inhibitors in metastatic colorectal cancer: a case report – Frontiers ↩︎
- ERS clinical practice guidelines on treatment of sarcoidosis | European Respiratory Society ↩︎
- Sarcoidosis: Evaluation and Treatment – AAFP ↩︎
- Treating sarcoidosis – European Lung Foundation ↩︎