La malattia di Erdheim-Chester è una patologia rara e complessa che rientra nel gruppo delle cosiddette istiocitosi non-Langerhans. In pratica, è causata da un accumulo anomalo di alcune cellule del sistema immunitario, chiamate istiociti, che invece di fare il loro lavoro normalmente, si infiltrano in vari organi e tessuti, causando infiammazione e danni. È una malattia multisistemica, il che significa che può colpire più parti del corpo, dalle ossa al cuore, dai polmoni al cervello.
La malattia prende il nome da due medici, Jakob Erdheim e William Chester, che la descrissero per la prima volta nel 1930. Per molti anni è rimasta una malattia poco conosciuta, ma grazie ai progressi della medicina e delle tecniche diagnostiche, oggi ne sappiamo molto di più.
Nonostante ciò, rimane una patologia rarissima, con solo poche centinaia di casi descritti in tutto il mondo. Si stima che colpisca circa 1 persona ogni 1-2 milioni. Di solito si manifesta in adulti tra i 50 e i 60 anni, con una leggera preferenza per gli uomini rispetto alle donne.
Eziopatogenesi

La causa esatta dell’ECD non è ancora del tutto chiara, ma sappiamo che è legata a alterazioni genetiche e disfunzioni del sistema immunitario. Alla base della malattia c’è un problema nelle cellule del sistema immunitario, in particolare gli istiociti, che invece di funzionare correttamente, iniziano a proliferare in modo incontrollato e a infiltrarsi in vari tessuti come ossa, polmoni, cuore, reni e sistema nervoso centrale.
In molti casi (circa il 50-60%), questo è dovuto a una mutazione genetica, la più comune delle quali è la BRAF V600E. Questa mutazione attiva in modo costante una via di segnalazione cellulare chiamata MAPK/ERK, che promuove la crescita e la sopravvivenza delle cellule. Altre mutazioni coinvolte includono quelle nei geni MAP2K1, PIK3CA, NRAS e ARAF, che agiscono su vie di segnalazione simili.
L’ECD è considerata una malattia infiammatoria cronica, in cui le cellule istiocitiche (macrofagi) si attivano in modo anomalo e rilasciano sostanze infiammatorie, come le citochine (es. IL-6, TNF-alfa). Questo processo infiammatorio contribuisce al danno tissutale e alla fibrosi (formazione di tessuto cicatriziale).

Clinica
L’infiltrazione e l’infiammazione provocati dalla malattia, come già detto, possono coinvolgere tutti i tessuti.
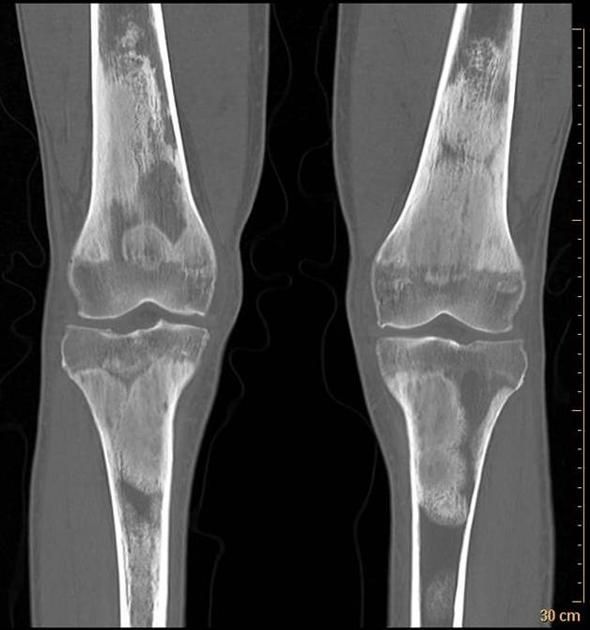
Il coinvolgimento osseo è presente in oltre il 95% dei casi ed è spesso il primo segno della malattia. I pazienti riferiscono dolore osseo, tipicamente localizzato alle regioni diafisarie e metafisarie delle ossa lunghe, come femori e tibie. Le radiografie mostrano lesioni osteosclerotiche (ispessimento osseo), spesso bilaterali e simmetriche, che rappresentano un marker diagnostico chiave.
Circa il 50% dei pazienti presenta manifestazioni neurologiche, che possono includere:
- atassia e disturbi dell’equilibrio;
- deficit cognitivi, come problemi di memoria o concentrazione;
- neuropatie periferiche, con formicolio o debolezza agli arti;
- disturbi visivi, tra cui diplopia o perdita della vista, dovuti all’infiltrazione delle strutture oculari o dei nervi ottici.
Il cuore e i grandi vasi sono colpiti nel 30-50% dei casi. Le manifestazioni includono pericardite, fibrosi miocardica e versamento pericardico, che possono causare dolore toracico e insufficienza cardiaca.
Il 20-40% dei pazienti sviluppa manifestazioni polmonari, come dispnea, soprattutto sotto sforzo e fibrosi polmonare, che riduce la capacità respiratoria e può progredire verso l’insufficienza respiratoria.
La fibrosi retroperitoneale, presente nel 30% dei casi, è una delle cause principali di coinvolgimento renale, con possibili complicanze come ostruzione ureterale e idronefrosi, con conseguente insufficienza renale.
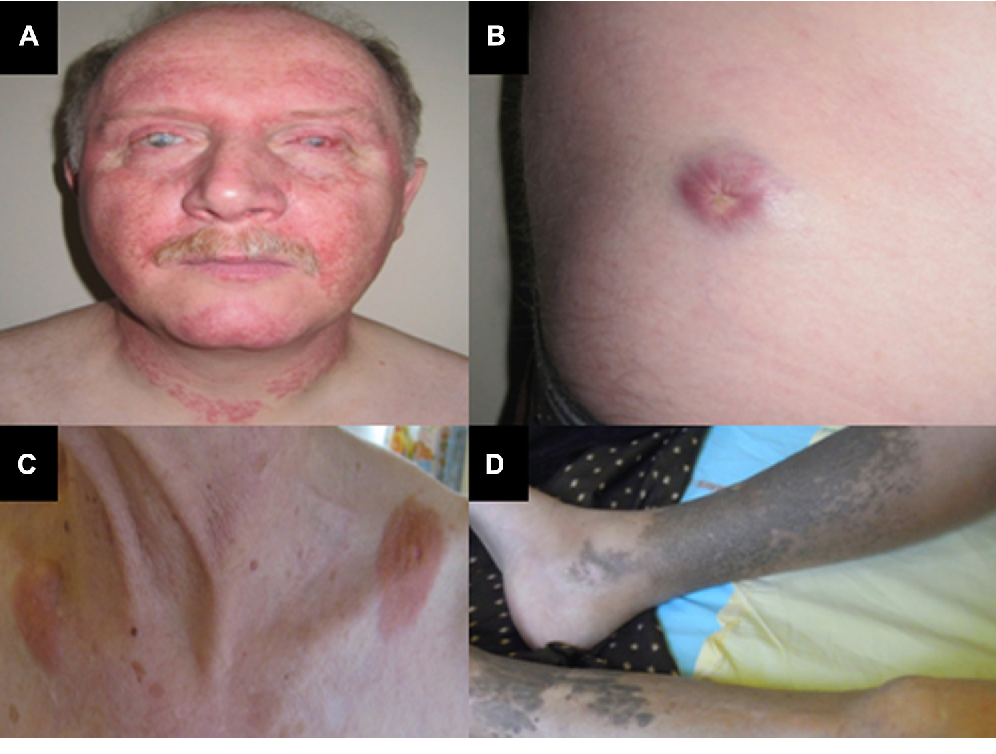
Circa il 25% dei pazienti presenta lesioni cutanee, come xantomi o noduli. Inoltre, l’ECD può colpire le ghiandole endocrine, causando diabete insipido (dovuto all’infiltrazione dell’ipofisi) o ipotiroidismo.
Oltre alle manifestazioni specifiche, molti pazienti presentano sintomi sistemici aspecifici, come:
- affaticamento cronico;
- febbre di origine sconosciuta;
- perdita di peso non intenzionale;
- dolori muscolari e articolari.
Questi sintomi, sebbene comuni a molte altre patologie, possono rappresentare un primo segnale della malattia.

Diagnosi
Diagnosticare l’ECD non è semplice, perché i sintomi possono essere confusi con quelli di altre malattie. Serve un approccio multidisciplinare e l’integrazione di dati clinici, radiologici, istologici e genetici. La diagnosi è spesso ritardata a causa della rarità della malattia e della sua presentazione clinica eterogenea.
La diagnosi inizia con una accurata anamnesi e un esame obiettivo. I sintomi più comuni che possono far sospettare l’ECD includono:
- dolore osseo (soprattutto alle gambe);
- sintomi neurologici (mal di testa, atassia, deficit cognitivi);
- manifestazioni sistemiche (affaticamento, febbre, perdita di peso);
- segni di coinvolgimento d’organo (dispnea, dolore toracico, insufficienza renale).
Tuttavia, poiché questi sintomi sono aspecifici, è necessaria una valutazione più approfondita.
L’imaging è uno strumento fondamentale per la diagnosi. Le tecniche più utilizzate includono:
- Rx, in particolare delle ossa lunghe (soprattutto femori e tibie), mostrano tipiche lesioni osteosclerotiche (ispessimento osseo), spesso bilaterali e simmetriche. Queste lesioni sono un marker diagnostico chiave;
- la TC è utile per valutare il coinvolgimento di organi come polmoni, reni e cuore. Ad esempio, può evidenziare fibrosi retroperitoneale, ispessimento pericardico o lesioni polmonari;
- la RM è particolarmente utile per valutare il coinvolgimento del sistema nervoso centrale (SNC). Può mostrare lesioni infiltrative nel cervello, nel midollo spinale o nei nervi ottici;
- La PET con FDG (fluorodesossiglucosio) è uno strumento prezioso per valutare l’estensione della malattia, poiché le cellule istiocitiche mostrano un elevato metabolismo glucidico. La PET può identificare lesioni attive in vari organi, guidando la scelta del sito per la biopsia.
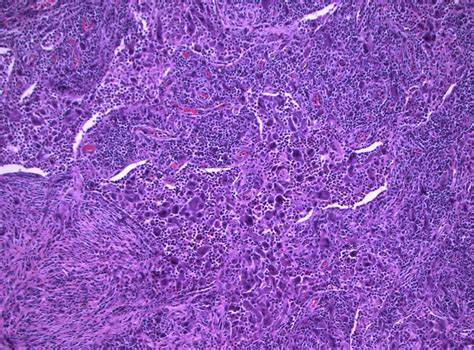
La conferma diagnostica richiede una biopsia del tessuto coinvolto (ad esempio, osso, pelle, tessuto retroperitoneale o polmone). L’analisi istologica rivela la presenza di: cellule istiocitiche con citoplasma schiumoso (foamy), dovuto all’accumulo di lipidi, infiammazione cronica e fibrosi.
Le cellule istiocitiche dell’ECD mostrano positività per marcatori come CD68, CD163 e fattore XIIIa, ma sono negative per CD1a (a differenza della malattia di Langerhans).
La ricerca di mutazioni genetiche è diventata parte integrante della diagnosi e della gestione dell’ECD. Le mutazioni più comuni includono: BRAF V600E, MAP2K1, PIK3CA, NRAS. L’identificazione di queste mutazioni non solo conferma la diagnosi, ma guida anche la scelta del trattamento (ad esempio, l’uso di inibitori di BRAF o MEK).
L’ECD deve essere distinta da altre patologie che possono presentarsi con sintomi simili, tra cui:
- malattia di Langerhans (un’altra forma di istiocitosi);
- linfomi o metastasi ossee;
- sarcoidosi;
- fibrosi retroperitoneale idiopatica;
- malattie infiammatorie croniche (es. vasculiti).
La combinazione di dati clinici, radiologici, istologici e genetici è essenziale per una diagnosi accurata.
Caricamento…
Trattamento
Il trattamento dipende dagli organi coinvolti e dalla gravità della malattia. Negli ultimi anni, grazie alla ricerca, sono state sviluppate terapie più mirate e personalizzate.
Le terapie mirate rappresentano il gold standard per il trattamento dell’ECD, soprattutto nei pazienti con mutazioni genetiche specifiche. Tra i principali farmaci rientrano:
- gli inibitori di BRAF, tra cui Vemurafenib e Dabrafenib, particolarmente efficaci nei pazienti con la mutazione BRAF V600E (presente in circa il 50-60% dei casi). Bloccano la via di segnalazione MAPK/ERK, riducendo la proliferazione delle cellule istiocitiche.
Studi clinici hanno dimostrato una significativa riduzione delle lesioni e un miglioramento dei sintomi in molti pazienti trattati con inibitori di BRAF; - gli inibitori di MEK, come Trametinib e Cobimetinib, utilizzati nei pazienti con mutazioni in MAP2K1 o in quelli che non tollerano gli inibitori di BRAF. Agiscono a valle della via MAPK/ERK, bloccando la proliferazione cellulare.
Gli inibitori di MEK hanno mostrato efficacia in pazienti con ECD, anche in assenza della mutazione BRAF V600E.
L’immunoterapia è stata a lungo il trattamento di prima linea per l’ECD, soprattutto prima dell’avvento delle terapie mirate.
L’interferone-alfa (IFN-α) modula il sistema immunitario, riducendo la proliferazione delle cellule istiocitiche e l’infiammazione. E’ stato ampiamente utilizzato con risultati variabili, ma rimane un’opzione per i pazienti senza mutazioni BRAF o MEK nonostante può dare affaticamento, sintomi simil-influenzali, depressione.
I corticosteroidi (es. prednisone) riducono l’infiammazione e i sintomi sistemici. Spesso usati in combinazione con altre terapie o per il controllo sintomatico a breve termine. L’uso prolungato è associato a effetti collaterali significativi (es. osteoporosi, iperglicemia).
In alcuni casi, soprattutto nei pazienti con malattia aggressiva o refrattaria, possono essere utilizzati farmaci chemioterapici tra cui:
- cladribina, un analogo delle purine che induce la morte cellulare (apoptosi) delle cellule istiocitiche, con risultati promettenti in alcuni pazienti con ECD, ma provocando immunosoppressione ed aumento del rischio di infezioni;
- vincristina o Citarabina, utilizzati in casi selezionati, soprattutto in combinazione con altri farmaci.
La ricerca continua a esplorare nuove opzioni terapeutiche per l’ECD, tra cui:
- inibitori di PI3K/AKT/mTOR: per i pazienti con mutazioni in PIK3CA o attivazione della via PI3K;
- terapie anti-citochine: farmaci che bloccano citochine pro-infiammatorie come l’IL-1 o il TNF-alfa.
- immunoterapie avanzate: come gli inibitori dei checkpoint immunitari (es. anti-PD1/PD-L1), ancora in fase di studio.
Oltre al trattamento specifico della malattia, è essenziale gestire le complicanze legate al coinvolgimento d’organo:
- supporto cardiaco: per pazienti con pericardite o insufficienza cardiaca;
- terapia renale: dialisi o gestione dell’idronefrosi nei casi di coinvolgimento renale;
- supporto respiratorio: ossigenoterapia o trattamento della fibrosi polmonare;
- terapia del dolore: per il controllo del dolore osseo o neuropatico.
Now loading…
Monitoraggio, Follow-up e Prognosi
Il monitoraggio regolare è fondamentale per valutare la risposta al trattamento e identificare eventuali recidive. Gli strumenti di monitoraggio includono:
Caricamento…
- imaging radiologico: TC, RM o PET per valutare la riduzione delle lesioni;
- esami di laboratorio: per monitorare la funzionalità d’organo (es. funzionalità renale, marker infiammatori);
- valutazione clinica: per identificare nuovi sintomi o complicanze.
La prognosi dell’ECD è migliorata significativamente con l’avvento delle terapie mirate. Tuttavia, dipende da:
- estensione della malattia: i pazienti con coinvolgimento del sistema nervoso centrale o cardiaco hanno una prognosi più severa;
- risposta al trattamento: i pazienti con mutazioni BRAF V600E rispondono meglio agli inibitori di BRAF;
- complicanze d’organo: il coinvolgimento di organi vitali può peggiorare la prognosi.
Prima delle terapie mirate, la sopravvivenza media era di 3-5 anni, ma oggi molti pazienti vivono più a lungo e con una migliore qualità di vita.
Fonti:
- Hematology: Basic Principles and Practice;
- Histiocytic Disorders;
- Erdheim-Chester disease – Orphanet;
- Erdheim-Chester disease – National Institutes of Health;
- Erdheim-Chester Disease Global Alliance (ECDGA);
- Erdheim-Chester disease – Radiopaedia;
- Erdheim-Chester disease: a comprehensive review – PubMed;
- Erdheim-Chester disease: consensus recommendations for evaluation, diagnosis, and treatment in the molecular era – PubMed.