
La sindrome da microdelezione 22q11.2, nota anche come sindrome velo-cardio-facciale o sindrome di DiGeorge, è la più comune tra le sindromi da microdelezione, con un’incidenza di circa 1 su 4.000 nati vivi.
Genotipo
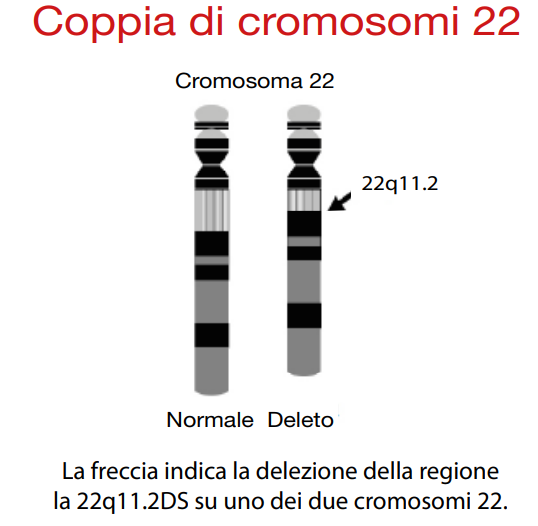
Questa condizione è caratterizzata dalla delezione di una porzione del cromosoma 22, che coinvolge tipicamente una regione di 3 megabasi (Mb) nel 95% dei casi e di 1,5 Mb nel 3%. Nel restante 2% dei pazienti, la delezione è atipica ma localizzata nella stessa area cromosomica. La presenza di regioni a bassa copia ripetuta (LCR) facilita questi riarrangiamenti cromosomici, contribuendo all’origine della delezione.
Nel 90-95% dei casi la microdelezione del cromosoma 22 è de novo e quindi non ereditaria.
In una piccola percentuale di casi la delezione si trasmette da uno dei genitori con meccanismo autosomico dominante: se uno dei genitori è portatore della delezione, ogni figlio avrà il 50% di probabilità di esserne affetto.
All’interno della regione eliminata sono presenti almeno 30 geni, tra cui il gene TBX1, la cui aploinsufficienza è ritenuta responsabile di molte delle manifestazioni cliniche osservate. Mutazioni puntiformi nel gene TBX1 sono state riscontrate in pazienti che presentavano difetti cardiaci tipici della sindrome, caratteristiche facciali peculiari e un variabile grado di ritardo psicomotorio.

Fenotipo
La sindrome di DiGeorge è una condizione genetica che si manifesta in modi molto diversi da persona a persona, persino all’interno della stessa famiglia. Le caratteristiche più comuni includono difetti cardiaci, tratti facciali distintivi, problemi al palato e difficoltà di apprendimento.
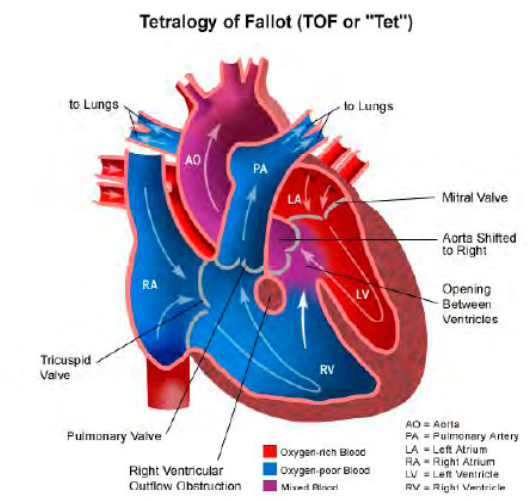
Una delle manifestazioni più frequenti è la presenza di cardiopatie congenite, riscontrate in oltre il 75% dei soggetti affetti e rappresentano il principale criterio diagnostico. Queste possono variare da forme più comuni come la tetralogia di Fallot, il tronco arterioso comune e l’arco aortico interrotto, a difetti del setto interatriale o interventricolare. Queste anomalie cardiache sono spesso accompagnate da particolari tratti del volto.
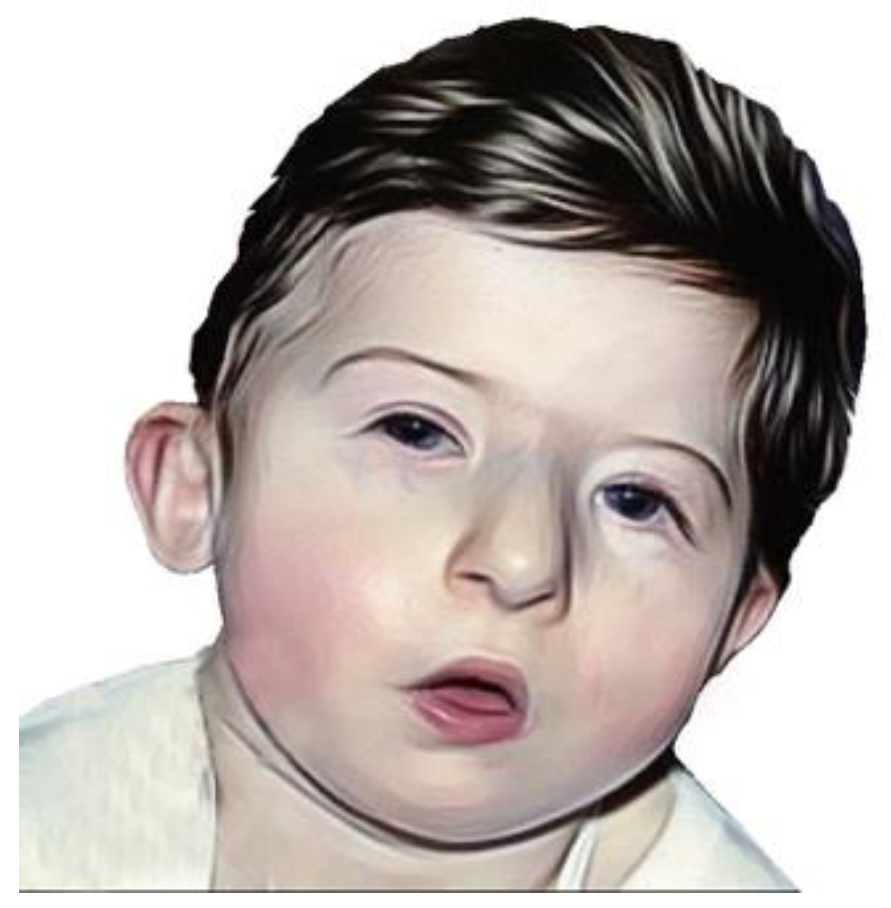
I tratti facciali tipici includono una bocca piccola, fessure palpebrali strette con ptosi palpebrale ed imbibimento edematoso periorbitario, padiglioni auricolari piccoli e a basso impianto, e un naso prominente con punta bulbosa e ali ipoplasiche. Queste caratteristiche possono attenuarsi con l’età, ma sono spesso presenti fin dalla nascita.
Circa il 70% dei pazienti presenta anomalie del palato, come l’insufficienza velo-faringea, che conferisce alla voce un timbro nasale caratteristico, o la palatoschisi, che può essere evidente o submucosa e quindi rilevabile solo attraverso un esame approfondito.
Molti bambini con questa sindrome affrontano sfide nell’apprendimento, con ritardi nello sviluppo psicomotorio che possono variare da lievi a più significativi. In alcuni casi, possono manifestarsi disturbi psicotici in età giovanile o adulta.

Altre possibili manifestazioni includono ipocalcemia neonatale transitoria dovuta ad ipoparatiroidismo, che può causare convulsioni ma tende a risolversi spontaneamente, e anomalie urogenitali come agenesia renale monolaterale, rene doppio, anomalie ostruttive delle vie escretrici o reflusso vescico-ureterale.
Le anomalie immunologiche sono frequenti, con deficit dell’immunità cellulo-mediata dovuti a ipoplasia o aplasia del timo.
Più frequentemente rispetto alla popolazione generale si osservano disturbi su base autoimmune quali artrite reumatoide giovanile, spesso poliarticolare, porpora trombocitopenica idiopatica, neutropenia autoimmune e malattia celiaca. Meno frequentemente sono stati descritti casi di anemia emolitica, pancitopenia autoimmune, tireopatia autoimmune (malattia di Graves) e diabete mellito.
Manifestazioni più rare sono: anomalie della corteccia cerebrale in forma di polimicrogiria, verosimilmente
secondaria a un danno vascolare, ano imperforato o anteriorizzato e coloboma dell’iride.

Diagnosi
La diagnosi di questa sindrome si basa inizialmente sull’osservazione clinica di segni caratteristici, come malformazioni cardiache congenite, anomalie del palato, dismorfismi facciali e ipocalcemia neonatale. Per confermare la diagnosi, si ricorre a test genetici specifici. Tradizionalmente, l’ibridazione fluorescente in situ (FISH) è stata utilizzata per rilevare la delezione sul cromosoma 22. Tuttavia, tecniche più recenti come la microarray CGH (ibridazione genomica comparativa) offrono una maggiore risoluzione nella rilevazione di microdelezioni. In alcuni casi, la diagnosi può essere sospettata già in epoca prenatale attraverso ecografie che evidenziano anomalie fetali; in tali situazioni, test prenatali non invasivi (NIPT) possono essere utilizzati per rilevare la delezione 22q11.2.

Trattamento
Non esiste una cura definitiva per la sindrome da microdelezione 22q11.2; pertanto, la gestione si concentra sul trattamento delle manifestazioni cliniche specifiche di ciascun paziente. Un approccio multidisciplinare è fondamentale e può includere:
- cardiopatie congenite: le malformazioni cardiache possono richiedere interventi chirurgici correttivi o altre procedure mediche, a seconda della gravità e del tipo di difetto presente;
- anomalie del palato: le anomalie come la palatoschisi possono necessitare di interventi chirurgici per migliorare la funzione del palato e correggere eventuali problemi di alimentazione o linguaggio;
- deficit immunitari: i pazienti con compromissione del sistema immunitario dovrebbero essere monitorati attentamente per prevenire e gestire infezioni ricorrenti. In alcuni casi, può essere indicata una terapia sostitutiva con immunoglobuline;
- ipocalcemia: i livelli di calcio devono essere monitorati regolarmente, soprattutto nei neonati, e l’ipocalcemia deve essere trattata con supplementi di calcio e vitamina D per prevenire complicanze come convulsioni;
- supporto allo sviluppo: programmi di intervento precoce, terapie del linguaggio, fisioterapia e supporto educativo possono aiutare a gestire le difficoltà di apprendimento e i ritardi nello sviluppo psicomotorio.
È essenziale che i pazienti siano seguiti da un team di specialisti, tra cui cardiologi, immunologi, endocrinologi, chirurghi plastici, logopedisti e neuropsichiatri infantili, per garantire una gestione completa e personalizzata della sindrome.
La prognosi varia ampiamente in base alla gravità delle manifestazioni cliniche e alla tempestività degli interventi terapeutici. Con una diagnosi precoce e un approccio terapeutico adeguato, molti individui con la sindrome da microdelezione 22q11.2 possono condurre una vita relativamente normale.
Fonti:
