Le anemie emolitiche congenite da difetti della membrana eritrocitaria rappresentano un gruppo eterogeneo di disordini rari, ma clinicamente rilevanti, caratterizzati da un’aumentata fragilità dei globuli rossi e dalla loro conseguente distruzione prematura. Queste patologie derivano da anomalie genetiche che compromettono la struttura e/o la funzione delle proteine della membrana eritrocitaria, determinando una morfologia cellulare alterata e una ridotta resistenza meccanica.
Tra le forme più rappresentative figurano la sferocitosi ereditaria, l’ellissocitosi ereditaria e la stomatocitosi ereditaria, ciascuna con peculiari caratteristiche morfologiche, genetiche e cliniche.
Sferocitosi ereditaria (malattia di Minkowski-Chauffard)
La sferocitosi ereditaria (HS, dall’inglese Hereditary Spherocytosis) è la più comune forma di anemia emolitica cronica congenita nella popolazione caucasica. L’incidenza è stimata in 1 caso ogni 2.000-5.000 individui, rendendola la principale causa di anemia emolitica congenita da difetto della membrana eritrocitaria.
La malattia fu descritta per la prima volta da Minkowski nel 1900 e successivamente approfondita da Chauffard, da cui il nome alternativo “malattia di Minkowski-Chauffard”.
Meccanismi patogenetici e basi molecolari
Alla base della sferocitosi ereditaria si riscontrano alterazioni genetiche che interessano le proteine strutturali del citoscheletro eritrocitario. Queste mutazioni compromettono l’architettura complessa della membrana cellulare, fondamentale per mantenere la caratteristica forma biconcava e la deformabilità dei globuli rossi.
Le principali proteine coinvolte includono la spectrina (nelle sue subunità α e β), l’ancherina (o banda 2.1), la banda 3 (scambiatore anionico 1) e la proteina 4.2. Queste componenti proteiche normalmente formano una rete strutturale che garantisce stabilità meccanica e flessibilità alla membrana eritrocitaria.
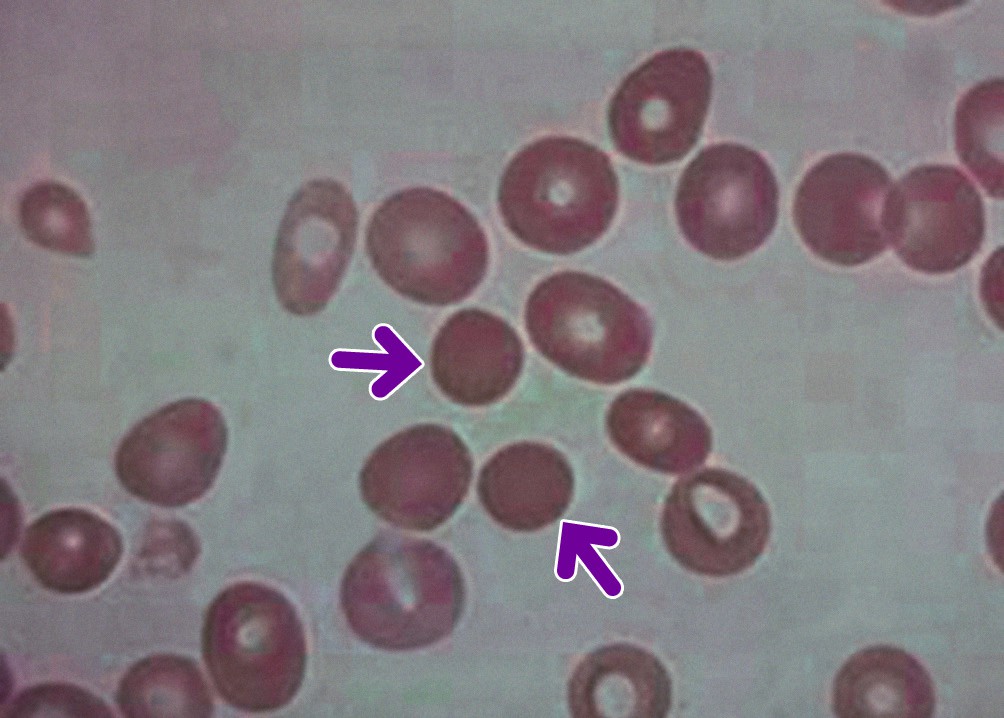
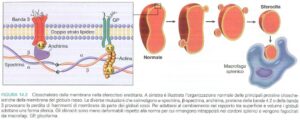
Le mutazioni genetiche determinano una progressiva perdita di frammenti di membrana, con conseguente riduzione del rapporto superficie/volume degli eritrociti. Questo processo trasforma gradualmente i normali discociti biconcavi in sferociti rigidi e meno deformabili.
Parallelamente, si instaura un’alterazione del bilancio ionico intracellulare, con aumento del consumo energetico per mantenere l’omeostasi elettrolitica. La disidratazione cellulare che ne consegue contribuisce ulteriormente alla rigidità degli eritrociti e all’aumento della concentrazione emoglobinica corpuscolare media (MCHC).
I globuli rossi modificati, privi della necessaria elasticità, incontrano difficoltà crescenti nel transitare attraverso i stretti sinusoidi splenici. La milza, agendo come filtro selettivo, riconosce e sequestra queste cellule anomale, avviando il processo di emolisi extravascolare.
Quadro clinico e variabilità di presentazione
La manifestazione clinica della sferocitosi ereditaria mostra un’ampia variabilità interindividuale, spaziando da forme completamente asintomatiche a quadri di anemia emolitica severa. Questa eterogeneità dipende dal tipo e dalla gravità delle mutazioni sottostanti, nonché da fattori epigenetici e ambientali.
Caricamento…
I pazienti affetti presentano tipicamente la triade classica dell’emolisi cronica: ittero (soprattutto evidente a livello sclerale), splenomegalia e colelitiasi pigmentaria. L’ittero risulta particolarmente evidente nei periodi neonatali e durante le crisi emolitiche.
La splenomegalia, presente nella maggior parte dei casi, riflette l’iperattività del sistema reticolo-endoteliale nel sequestrare e distruggere gli eritrociti anomali. L’ingrossamento splenico può diventare clinicamente evidente già nell’infanzia.
Tra le complicanze più rilevanti si annoverano le crisi emolitiche acute, spesso scatenate da infezioni intercorrenti. Particolarmente temibile è la crisi aplastica da infezione da Parvovirus B19, che può determinare una grave soppressione dell’eritropoiesi.
Le alterazioni scheletriche, come la prominenza frontale o la polidattilia, rappresentano manifestazioni meno comuni ma caratteristiche, conseguenti all’espansione del midollo emopoietico. Le ulcere malleolari, sebbene rare, costituiscono un segno distintivo della malattia avanzata.
Approccio diagnostico
La diagnosi di sferocitosi ereditaria si avvale di un iter articolato che combina dati clinici, laboratoristici e strumentali. L’esame emocromocitometrico rivela tipicamente un’anemia normocitica o lievemente microcitica, con aumento caratteristico della MCHC.
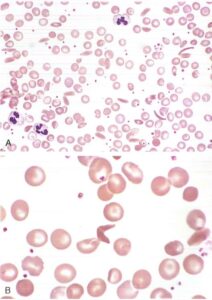
Lo striscio di sangue periferico rappresenta un esame fondamentale, mostrando la presenza di sferociti. Tuttavia, è importante ricordare che questi elementi cellulari non sono patognomonici, essendo riscontrabili anche in altre condizioni emolitiche.
N.B. In alcune anemie immunoemolitiche da IgG è possibile osservare anche sferociti. Questo accade perché, nelle anemie emolitiche estrinseche mediate da anticorpi, i globuli rossi opsonizzati non vengono totalmente distrutti al primo passaggio attraverso la milza, ma vengono eliminate solo alcune porzioni di membrana. Questo processo induce nei globuli rossi una forma sferica, che li rende meno deformabili e più suscettibili alla rimozione. Tali cellule vengono poi totalmente distrutte durante il secondo passaggio nella milza.
Il test di fragilità osmotica mantiene ancora un ruolo diagnostico, dimostrando la maggiore suscettibilità degli sferociti alla lisi in ambienti ipotonici. Gli sferociti si gonfiano per accumulo di acqua e si rompono più precocemente rispetto ai globuli rossi normali. Questo fenomeno può essere prevenuto con l’aggiunta di glucosio.
L’analisi dell’emocromo con parametri avanzati (come il conteggio dei reticolociti) completa il quadro laboratoristico.
L’avvento delle tecniche di biologia molecolare ha rivoluzionato l’approccio diagnostico. L’analisi genetica, disponibile presso centri specializzati, permette oggi di identificare con precisione le mutazioni responsabili, offrendo anche possibilità di consulenza genetica familiare.
Strategie terapeutiche
La gestione della sferocitosi ereditaria richiede un approccio personalizzato, modulato in base alla severità del quadro clinico e alla presenza di complicanze. L’osservazione clinica periodica rappresenta la pietra angolare della gestione dei casi lievi.

L’intervento di splenectomia rimane l’opzione terapeutica più efficace nei casi sintomatici, determinando nella maggior parte dei pazienti una significativa riduzione dell’emolisi. Tuttavia, la decisione chirurgica deve essere ponderata attentamente, considerando i rischi infettivi e trombotici a lungo termine.
La tempistica ideale per l’intervento viene generalmente posticipata oltre i 5-6 anni di età, previa adeguata copertura vaccinale (antipneumococcica ed antihaemophilus). La profilassi antibiotica post-splenectomia, particolarmente cruciale in età pediatrica, rappresenta un caposaldo della gestione post-operatoria.
La supplementazione con acido folico viene raccomandata in tutti i pazienti, al fine di sostenere l’aumentato turnover eritrocitario e prevenire le crisi megaloblastiche. Nei casi più gravi, può rendersi necessaria una terapia trasfusionale periodica.
La ricerca attuale sta esplorando nuove strategie terapeutiche, tra cui l’utilizzo di inibitori del complemento e approcci di terapia genica. Queste innovazioni potrebbero in futuro offrire alternative alla splenectomia, particolarmente per i pazienti più giovani.
Monitoraggio e follow-up
I pazienti affetti da sferocitosi ereditaria richiedono un follow-up ematologico regolare, volto a identificare precocemente eventuali complicanze. Il monitoraggio ecografico della colecisti e la valutazione periodica della funzionalità epatica costituiscono elementi essenziali del percorso assistenziale.

Ellissocitosi ereditaria (HE)
L’ellissocitosi ereditaria (HE) è una malattia autosomica dominante caratterizzata da un’alterazione della spettrina, una proteina fondamentale per la stabilità strutturale della membrana eritrocitaria. Mutazioni nei geni SPTA1, SPTB (codificanti per le subunità α e β della spettrina) o EPB41 (proteina 4.1) compromettono l’interazione tra citoscheletro e membrana, portando alla formazione di globuli rossi di forma ellittica o ovale (ellissociti).
Clinica
Presenta un’ampia variabilità fenotipica:
- forme asintomatiche (70-80% dei casi): i pazienti non sviluppano anemia significativa e la diagnosi è spesso occasionale, rilevata tramite esami del sangue di routine;
- forme sintomatiche: possono manifestarsi con anemia emolitica lieve-moderata, ittero intermittente, splenomegalia e, raramente, colelitiasi da bilirubina. Le crisi emolitiche acute sono scatenate da infezioni (es. parvovirus B19) o stress ossidativo;
- variante grave (piropoichilocitosi ereditaria, HPP): anemia trasfusione-dipendente, ritardo di crescita e complicanze scheletriche (fronte prominente).
Diagnosi

- striscio di sangue periferico: presenza di ellissociti (>60% dei globuli rossi), talvolta associati a ovalociti o frammenti cellulari;
- test di fragilità osmotica: nella maggior parte dei casi è normale, a differenza della sferocitosi ereditaria. Diventa patologico solo nelle forme severe (HPP);
- test specializzati:
- ectacitometria: valuta la deformabilità eritrocitaria, ridotta nell’HE;
- analisi SDS-PAGE: identifica deficit quantitativi/qualitativi delle proteine di membrana (spettrina, proteina 4.1);
- test genetici: confermano le mutazioni in SPTA1, SPTB o EPB41, utili per la consulenza familiare.
L’elissocitosi ereditaria entra in diagnosi differenziale con:
- sferocitosi ereditaria: globuli rossi sferici, fragilità osmotica aumentata, anemia più frequente;
- anemie emolitiche autoimmuni: test di Coombs positivo, assenza di ellissociti;
- deficit di piruvato chinasi: anemia con poichilociti, test enzimatici specifici.
Trattamento e gestione
- forme asintomatiche: nessuna terapia necessaria; monitoraggio periodico;
- forme sintomatiche:
- acido folico (1-5 mg/die): supporta l’eritropoiesi compensatoria;
- trasfusioni: indicate solo nelle crisi emolitiche gravi o nell’HPP;
- splenectomia: riservata ai casi con anemia emolitica severa, preferibilmente dopo i 5 anni per ridurre il rischio infettivo. Migliora l’emolisi ma non corregge la morfologia eritrocitaria.
Nella maggior parte dei casi, l’HE ha un decorso benigno e non influisce sull’aspettativa di vita. Le forme gravi (HPP) richiedono un follow-up multidisciplinare per gestire le complicanze.
Now loading…
Stomatocitosi congenite
Le stomatocitosi congenite rappresentano un gruppo eterogeneo di disordini rari dell’eritropoiesi, caratterizzati da un’alterata permeabilità della membrana eritrocitaria agli elettroliti, in particolare ai cationi come sodio (Na⁺) e potassio (K⁺). A differenza delle altre forme di anemia emolitica da difetti della membrana, la patogenesi della stomatocitosi non risiede in una debolezza strutturale dello scheletro della membrana, bensì in un’anomala regolazione del trasporto ionico transmembrana.
Queste alterazioni determinano un disequilibrio osmotico che provoca un accumulo intracellulare di sodio e acqua, con conseguente rigonfiamento dei globuli rossi.
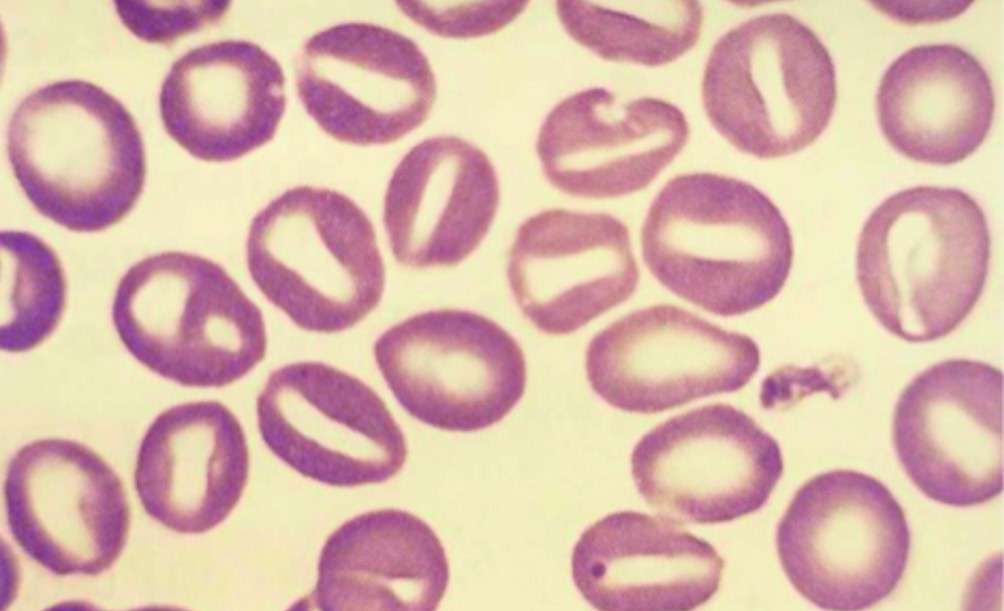
Morfologicamente, gli eritrociti appaiono stomatocitici, ossia presentano un’area centrale di pallore dalla forma allungata o “a fessura”, simile a una bocca (da cui il termine stoma, cioè “bocca” in greco), invece del classico aspetto rotondeggiante del centro pallido dei normali globuli rossi.
La stomatocitosi ereditaria può manifestarsi in forme cliniche variabili, dalla quasi completa asintomaticità a quadri di anemia emolitica cronica moderata o severa.
Viene classificata in base allo stato di idratazione dei globuli rossi. Le due varianti principali sono quella sovraidratata (idrocitosi) e quella deidratata (xerocitosi). Nel primo caso l’MCHC risulterà basso, nel secondo caso elevato.

La diagnosi si avvale della valutazione morfologica su striscio periferico (con presenza di stomatociti in più del 10% dei globuli rossi), test di fragilità osmotica, analisi del contenuto elettrolitico intraeritrocitario e, nei casi dubbi o per fini diagnostici avanzati, studi genetici mirati.
Il trattamento è generalmente di supporto, e la splenectomia, a differenza della sferocitosi, non è raccomandata poiché può aumentare il rischio di trombosi, specialmente nelle forme con disidratazione eritrocitaria.
Altre fonti:
- Orphanet: ellissocitosi ereditaria;
- MSD Manuals: sferocitosi ed ellissocitosi ereditaria;
- Medicover Hospitals: ellissocitosi ereditaria.
Caricamento…