Le alterazioni delle catene di globina rappresentano un capitolo importante nel contesto delle anemie emolitiche, ma è fondamentale sottolineare che il quadro clinico e fisiopatologico di queste condizioni è complesso e misto, includendo elementi sia di emolisi che di anemia iporigenerativa congenita. Le anomalie dell’emoglobina (Hb) sono causate da mutazioni genetiche che possono influenzare sia la quantità che la qualità delle catene globiniche.
Le principali alterazioni si distinguono in due grandi categorie:
- ridotta sintesi di una o più catene globiniche, come avviene nelle talassemie (a trasmissione autosomica recessiva);
- anomalie strutturali della globina, che danno origine alle cosiddette emoglobinopatie strutturali.
La composizione dell’emoglobina normale
Nel soggetto adulto sano, l’emoglobina è presente in diverse forme, costituite da tetrameri formati da quattro catene globiniche. Le principali frazioni sono:

- emoglobina A1 (HbA1): rappresenta circa il 97% dell’emoglobina totale ed è formata da due catene alfa (α) e due catene beta (β) → α₂β₂;
- emoglobina A2 (HbA2): costituisce circa il 2% del totale, con composizione α₂δ₂;
- emoglobina fetale (HbF): formata da due catene alfa e due catene gamma → α₂γ₂. Grazie alla sua maggiore affinità per l’ossigeno rispetto all’HbA, facilita il trasferimento di ossigeno dalla madre al feto. Dopo la nascita, la HbF viene progressivamente sostituita dalla HbA, e i suoi livelli si riducono fisiologicamente fino a stabilizzarsi attorno all’1% entro il sesto mese di vita.
Oltre alla componente proteina delle catene polipeptidiche, l’emoglobina presenta un gruppo eme, costituito da una protoporfirina IX legata a un atomo di ferro ferroso (Fe²⁺), che rappresenta il sito attivo per il legame reversibile con l’ossigeno.
Caricamento….
Diminuzione della sintesi delle catene di globina: le talassemie
Beta-Talassemia
Le talassemie beta rappresentano un gruppo di disordini genetici dovuti a mutazioni “loss of function” che interessano i geni delle catene della globina, la catena beta dell’emoglobina A (HbA). Tali mutazioni comportano difetti a vari livelli del processo di espressione genica: trascrizione, maturazione o traduzione dell’mRNA. Il risultato è una ridotta o assente produzione delle catene beta, con conseguente diminuzione della sintesi di HbA o, nei casi più gravi, una sua totale assenza.
Il deficit di sintesi di una catena porta a uno squilibrio tra le catene globiniche, con un eccesso relativo delle catene non coinvolte dalla mutazione. Queste ultime, non potendo assemblarsi in modo corretto, tendono a formare tetrameri anomali o a precipitare, generando tossicità cellulare, morte eritroide e, quindi, un’anemia che è il risultato sia di eritropoiesi inefficace che di emolisi.
In base alla gravità del difetto genetico e alla modalità di trasmissione, distinguiamo principalmente due forme cliniche: talassemia minor e talassemia major.
Talassemia minor (carattere talassemico)

Questa forma, anche detta eterozigote, è molto diffusa soprattutto nelle popolazioni dell’area mediterranea, compresa l’Italia. Generalmente si tratta di una condizione asintomatica, rilevata casualmente durante esami ematologici di routine.
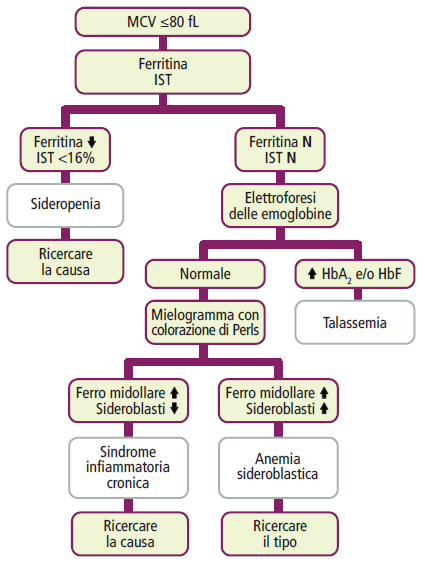
È causata da una diminuzione parziale della sintesi delle catene beta. Il quadro ematologico tipico mostra un numero di emazie normale o aumentato, ma con anemia lieve, microcitica (MCV < 65 fL) e ipocromica (MCHC ridotto). L’emoglobina totale si aggira generalmente tra 10,5 e 12 g/dL.
All’elettroforesi dell’emoglobina si osservano caratteristiche alterazioni:
- HbA1: leggermente ridotta;
- HbA2: aumentata lievemente;
- HbF: generalmente normale, ma può essere lievemente aumentata nel 50% dei casi.
Non richiede trattamento, ma è importante identificare i portatori, soprattutto in ambito di counseling genetico. In caso di coppia in cui entrambi i partner siano portatori del tratto talassemico, vi è un 25% di probabilità di avere un figlio sano, un 50% di probabilità che sia portatore (talassemia minor) e un 25% di probabilità che presenti la forma grave (talassemia major).
Nota clinica: si deve sospettare un carattere talassemico in caso di anemia marcata ma sproporzionatamente microcitica, con MCV anche inferiore a 60 fL. Le talassemie sono tra le forme di anemia più microcitiche in assoluto.
| Parametro | Talassemia Minor | Anemia Sideropenica |
|---|---|---|
| MCV (volume corpuscolare medio) | ↓↓↓ (molto basso) | ↓ (basso) |
| MCHC (concentrazione media di Hb nei GR) | Normale o leggermente ↓ | ↓↓↓ (marcatamente ridotto) |
| HbA₂ | ↑ (>3.5%) | Normale o ↓ |
| RDW (ampiezza di distribuzione eritrocitaria) | Normale | ↑ (anisocitosi marcata) |
| Ferritina sierica | Normale o ↑ | ↓↓↓ |
| Sideremia | Normale o ↑ | ↓↓↓ |
| Saturazione transferrina | Normale o ↑ | ↓↓↓ |

Talassemia major (anemia di Cooley)
La talassemia major, o forma omozigote, è una malattia grave causata dall’assenza totale della sintesi delle catene beta, con conseguente quasi totale assenza di HbA1 (presente solo in tracce, circa 1%) e marcato incremento di HbF (fino al 98% dell’emoglobina totale). Il tentativo del midollo di compensare l’anemia porta a un’attivazione massiva dell’eritropoiesi, che però risulta inefficace. I tetrameri formati da catene alfa libere, instabili e insolubili, precipitano all’interno delle emazie formando corpi di Heinz, analoghi a quelli osservati nel deficit di G6PD.
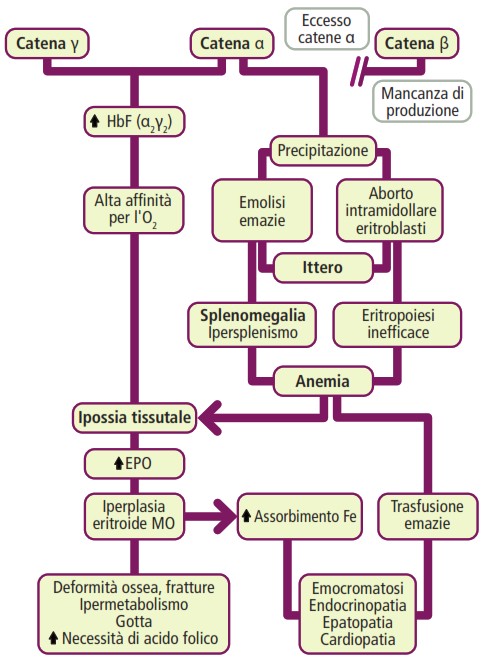
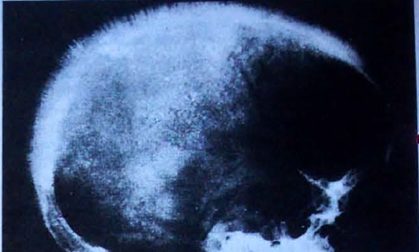
L’esordio clinico non si manifesta alla nascita, ma in genere tra i 6 e gli 8 mesi di vita, quando l’HbF fisiologica inizia a diminuire e dovrebbe essere sostituita dalla produzione di HbA. I sintomi comprendono anemia emolitica cronica severa (emolisi intramidollare), splenomegalia, epatomegalia variabile e caratteristiche alterazioni ossee, tra cui il tipico aspetto a spazzola del cranio e disformie facciali con impianto dentale anomalo.
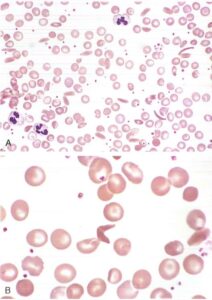
Nel sangue periferico si osservano:
- anisopoichilocitosi (variazione di forma e dimensioni delle emazie);
- ellissociti, dacriociti, emazie con punteggiatura basofila;
- eritroblasti circolanti;
- reticolocitosi, ma inferiore alle attese rispetto al grado di anemia.
All’elettroforesi dell’Hb si riscontra:
- HbA1: molto ridotta o assente;
- HbF: aumentata significativamente (60-98% di tutta l’Hb);
- HbA2: può essere normale o lievemente aumentata.
La gestione della talassemia major è tuttora prevalentemente palliativa. Il trattamento standard consiste in trasfusioni regolari, che però comportano il rischio di sovraccarico di ferro: per questo motivo è fondamentale l’uso di chelanti del ferro (es. deferoxamina, deferasirox).
In alcuni casi, soprattutto in presenza di ipersplenismo o splenomegalia massiva, può essere indicata la splenectomia.
Caricamento…
Esistono inoltre terapie farmacologiche che stimolano la produzione di HbF, come idrossiurea, butirrato e 5-azacitidina. Questi farmaci inducendo la produzione di catene gamma, aumentano l’HbF, che ha una maggiore stabilità e può migliorare la sopravvivenza eritrocitaria, con benefici sull’anemia.
Più recentemente, è stato introdotto luspatercept, un agente stimolante la maturazione eritroide, approvato per l’uso in pazienti adulti con beta-talassemia trasfusione-dipendente.
L’unica terapia curativa attualmente disponibile per la talassemia major è il trapianto allogenico di midollo osseo, che può portare alla guarigione definitiva nei pazienti compatibili. Un’alternativa in fase avanzata di sperimentazione è il trapianto autologo con editing genetico, volto alla correzione della mutazione del gene della catena beta, mediante tecniche di ingegneria genetica (es. CRISPR/Cas9).
Alla luce delle implicazioni cliniche e riproduttive, è fondamentale promuovere lo screening della popolazione, in particolare tra le donne in età fertile, per identificare i soggetti eterozigoti. Lo studio degli indici corpuscolari (MCV, MCHC) rappresenta un metodo semplice, efficace e poco costoso per il riconoscimento dei portatori, anche in assenza di sintomi. Una diagnosi precoce consente una corretta consulenza genetica, fondamentale per la prevenzione della talassemia major.
Alfa-talassemie
Le alfa-talassemie costituiscono il disordine genetico dell’emoglobina più diffuso al mondo, in particolare tra le popolazioni del sud-est asiatico, dell’Africa e del Medio Oriente. In Italia, al contrario, sono piuttosto rare. A differenza delle beta-talassemie, in cui è coinvolta la sintesi della catena beta, le alfa-talassemie derivano da una ridotta o assente produzione delle catene alfa della globina, essenziali per la formazione sia della HbA (α2β2) che della HbF (α2γ2).
Now loading…
Il gene che codifica per la catena alfa è duplicato su ciascun cromosoma 16, per un totale di quattro alleli. La gravità clinica è direttamente proporzionale al numero di geni inattivati da mutazioni “loss of function”, in genere delezioni.
- Portatore silente (delezione di un solo gene alfa)
In caso di perdita di un solo allele, la persona è completamente asintomatica. La diagnosi è spesso casuale o familiare, perché gli esami di routine risultano generalmente nella norma e l’elettroforesi dell’emoglobina non mostra alterazioni. - Alfa-talassemia minor (o tratto alfa-talassemico)
Con la perdita di due geni alfa, il quadro clinico resta in genere lieve o assente, ma si può riscontrare una lieve anemia microcitica. A differenza della beta-talassemia minor, però, l’elettroforesi dell’Hb è solitamente normale, il che può rendere più difficile il sospetto diagnostico.
Questa forma viene spesso confusa con altre anemie microcitiche (es. sideropenia), per cui è importante sospettarla in presenza di anemia lieve non spiegabile e non responsiva alla terapia marziale, soprattutto se in pazienti provenienti da aree endemiche. - Emoglobinopatia H (perdita di tre geni alfa)
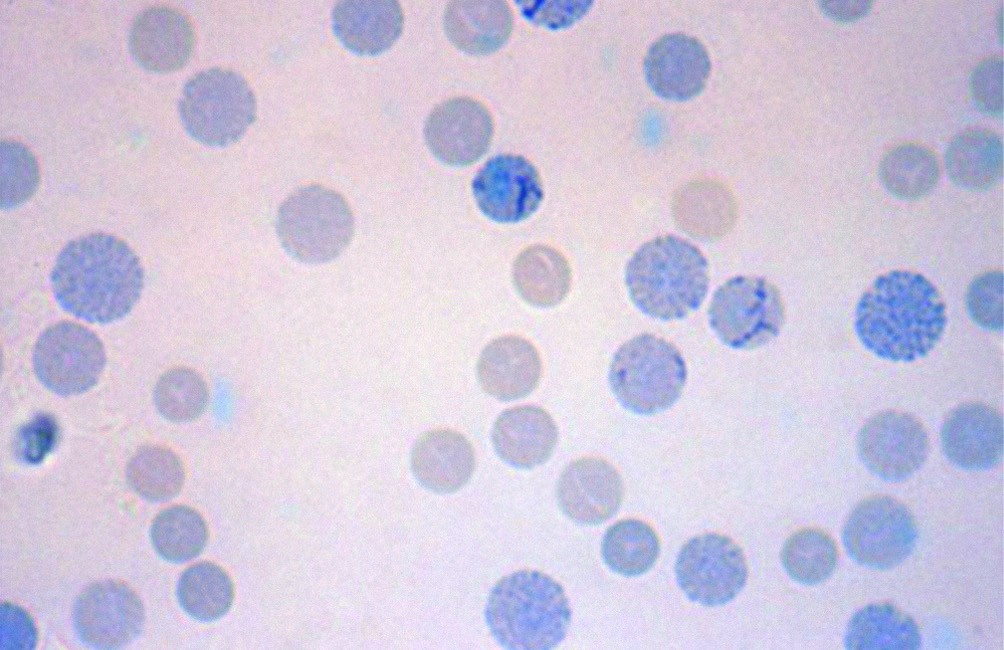
La forma intermedia dell’alfa-talassemia si manifesta quando solo un gene alfa funziona. In questa condizione, le catene beta, normalmente bilanciate dalle catene alfa, si accumulano e formano tetrameri instabili (HbH, β4). Questi precipitati intracellulari sono visibili come corpi di inclusione con colorazione brillante (tipico aspetto “a pallina da golf”).
Dal punto di vista clinico, l’emoglobinopatia H è caratterizzata da:- anemia emolitica cronica di grado variabile;
- splenomegalia;
- talora episodi di crisi emolitiche acute (ad esempio in corso di infezioni).
- Emoglobina di Bart (idrope fetale)
La perdita completa di tutti e quattro i geni alfa comporta una condizione estremamente grave: il feto è incapace di sintetizzare catene alfa, e quindi non può produrre né HbF né HbA. Si formano tetrameri esclusivi di catene gamma (Hb Bart, γ4), che non riescono a trasportare efficacemente l’ossigeno.
Il risultato è una forma letale di anemia intrauterina, nota come idrope fetale da Hb Bart, che conduce a morte in utero o neonatale precoce. In rare situazioni, la diagnosi prenatale consente un intervento intrauterino con trasfusioni, ma il decorso rimane generalmente infausto.
Il principio generale di trattamento delle alfa-talassemie gravi (come per la beta-talassemia major) è simile, con approcci palliativi nei casi meno severi e trapianto allogenico di midollo osseo nei pazienti con emoglobinopatia H sintomatica o grave.
Nei soggetti trasfusione-dipendenti, è fondamentale monitorare e gestire il sovraccarico di ferro, anche in questi casi meno frequenti. La possibilità di terapie geniche per le alfa-talassemie è al momento ancora più limitata rispetto alle beta-talassemie, ma in prospettiva futura potrebbe estendersi anche a questa forma.
Now loading…
Emoglobinopatie strutturali
Le emoglobinopatie strutturali rappresentano un gruppo eterogeneo di disordini genetici dovuti a mutazioni puntiformi nei geni delle catene globiniche, che determinano la produzione di emoglobine con struttura anomala. A differenza delle talassemie, in cui il difetto riguarda la quantità di globina sintetizzata, in questo caso il problema risiede nella qualità delle catene globiniche prodotte.
Queste mutazioni possono avere effetti diversi sulla funzione e stabilità della molecola di emoglobina, e per questo motivo si classificano in tre grandi categorie, in base alle conseguenze funzionali della mutazione:
Emoglobine instabili
In alcune varianti, la mutazione altera la stabilità molecolare dell’emoglobina, rendendola soggetta a denaturazione spontanea e formazione di precipitati intraeritrocitari. Queste inclusioni proteiche danneggiano la membrana dei globuli rossi e portano alla loro distruzione prematura, con conseguente anemia emolitica cronica.
Le emoglobine instabili sono tipicamente difficili da diagnosticare con elettroforesi convenzionale e richiedono test specifici (come il test dell’isopropanolo o del calore). Clinicamente, il quadro può simulare altre forme di anemia emolitica, anche se di solito è più lieve. Un esempio classico è l’emoglobina Köln.
Emoglobine con affinità alterata per l’ossigeno
Alcune varianti emoglobiniche mantengono una struttura stabile, ma presentano una modificata affinità per l’ossigeno. In questo gruppo si distinguono:
- emoglobine con aumentata affinità per l’ossigeno, che rilasciano meno ossigeno ai tessuti. Questo può simulare un quadro di policitemia secondaria (aumentata produzione di eritrociti per compensare l’ipossia tissutale percepita);
- emoglobine con affinità ridotta, che rilasciano l’ossigeno troppo facilmente, e possono determinare ipossia tissutale e cianosi persistente, anche in presenza di saturazioni arteriose normali.
Queste varianti non sono emolitiche, e spesso vengono scoperte casualmente in corso di approfondimenti per anomalie nei livelli di emoglobina o saturazione.
Emoglobine con solubilità alterata

La categoria più importante e frequente è rappresentata dalle emoglobinopatie con alterata solubilità, le cosiddette varianti S, C, D, E, etc., tra cui la più nota è l’emoglobina S (HbS), responsabile della drepanocitosi o anemia falciforme. In questo tipo di disordine, la mutazione induce una polimerizzazione dell’emoglobina deossigenata, che causa la deformazione dei globuli rossi in forme a falce (drepanociti), determinando:
- emolisi cronica
- occlusione vascolare
- ischemia tissutale
L’emoglobina C (HbC), invece, è meno grave ma può anch’essa associarsi a forme moderate di anemia emolitica, soprattutto in combinazione con HbS (malattia SC).
Queste emoglobinopatie sono particolarmente diffuse in Africa e nelle popolazioni afrodiscendenti, e la loro frequenza è aumentata in molte aree del mondo per via della migrazione.